



Туберозный склероз: этиология, клиническая манифестация, диагностика
Туберозный склероз (ТС) – мультисистемное генетически гетерогенное заболевание (аутосомно-доминантное, с неполной пенетрантностью и вариабельной экспрессивностью). Для него характерна высокая частота возникновения новых (спонтанных) мутаций, которые обнаруживаются в 68% всех случаев ТС, клинически дебютирующих в раннем возрасте. Учитывая системность поражения, клиническую манифестацию и существующие подходы к диагностике, часто используется термин «комплекс туберозного склероза».
Клинические симптомы
Это заболевание характеризуется широким спектром клинических проявлений с развитием доброкачественных неинвазивных опухолеподобных очагов поражения (гамартом), локализованных в тканях головного мозга, сердца, почек, легких, печени и кожи.
Распространенность ТС среди новорожденных составляет приблизительно 1 на 6000 вне зависимости от пола или расы. Некоторые исследования показали, что мужчины чаще имеют поражения центральной нервной системы (ЦНС), но это не было окончательно доказано. Симптомы ТС могут проявиться в любом возрасте. У новорожденных и детей до 7 лет ТС диагностируется при тяжелой эпилепсии, аутизме или сердечной недостаточности. У пожилых пациентов могут наблюдаться клинические варианты с поражением почек, легких либо кожи и слизистых оболочек.
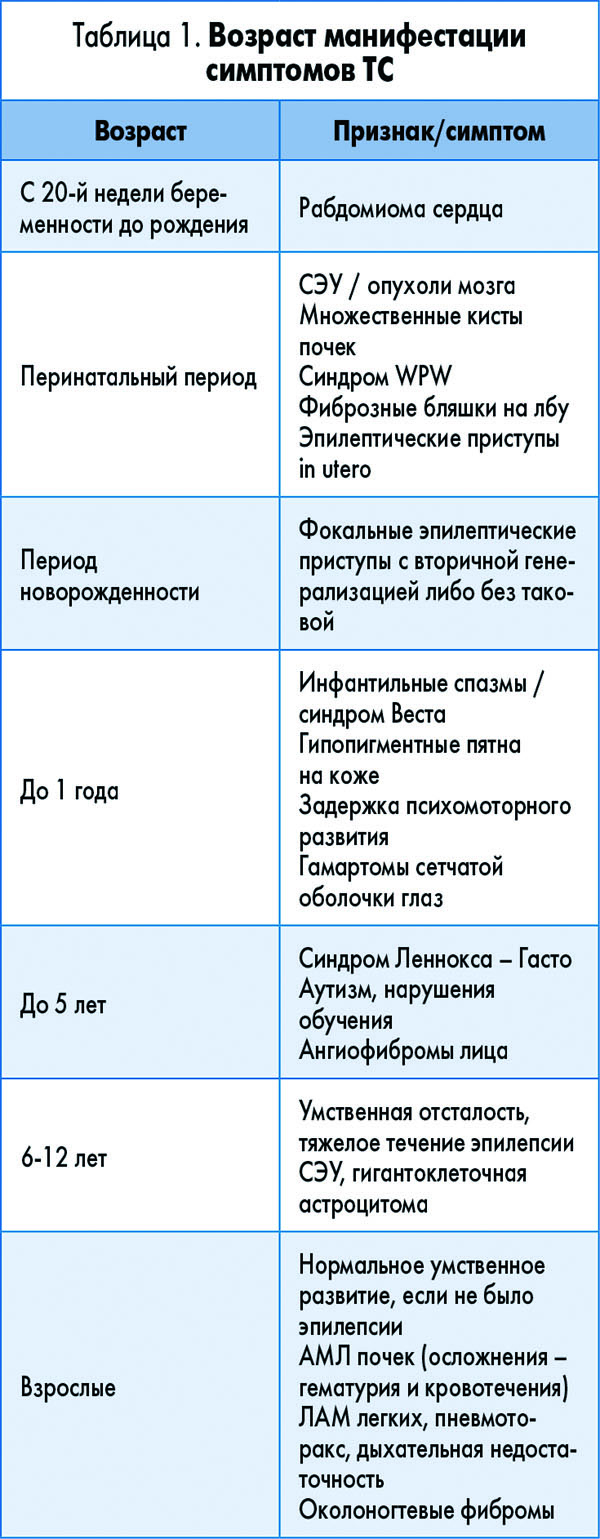
В зависимости от возраста пациента превалируют те или иные клинические варианты заболевания с локализацией поражения в разных системах органов.
• Поражение сердца происходит во время внутриутробного развития или в неонатальном периоде. Рабдомиома может регрессировать в течение долгого времени.
• Эпилепсия, задержки психического развития, аутизм манифестируют в раннем детстве до подросткового периода.
• Поликистоз почек, как правило, проявляется в младенчестве или раннем детстве.
• Ангиомиолипома (АМЛ) почек может быть диагностирована в любом возрасте.
• Лимфоангиомиоматоз (ЛАМ) легких обычно отмечается в третьем или четвертом десятилетии жизни пациента.
Точную оценку заболеваемости и распространенности ТС затрудняют недостаточная диагностика менее тяжелых фенотипов высокая спонтанная частота мутаций (примерно две трети всех случаев). Кроме того, на выявляемость заболевания влияет вариабельность симптоматики (даже в пределах известного семейного анамнеза пациентов), а также нежелание родителей/родственников, у которых симптомы отсутствуют, проходить генетическое тестирование. Разнообразие клинических признаков ТС, вариабельность фенотипа, тот факт, что манифестация проявлений заболевания зависит от возраста пациента, существенно затрудняют диагностику и требуют мультидисциплинарного подхода.
Практически всегда при ТС поражается ЦНС, вызывая инвалидизирующие неврологические нарушения. В их числе:
• эпилепсия, которая наблюдается у 90% пациентов, страдающих ТС;
• образование субэпендимальных узлов (СЭУ) – у 90-100%;
• субэпендимальные гигантоклеточные астроцитомы (СЭГА) – у 5-20%;
• замедление умственного развития (у 44-64%);
• микроструктурные аномалии белого вещества головного мозга.
Другими манифестациями ТС, не затрагивающими нервную систему, являются гипопигментные пятна и ангиофибромы лица, наличие кист почек и/или АМЛ, лимфангиолейомиомы легких, рабдомиомы сердца, гамартомы сетчатки и ангиомы печени. У 85% пациентов, страдающих ТС, выявляют мутации одного из двух генов, подавляющих рост опухолей, – TSC1 (кодирующего гамартин) или TSC2 (кодирующего туберин). Гамартин и туберин участвуют в регуляции пролиферации и дифференциации клеток. Они образуют физический и функциональный комплекс, активирующий гуанозин трифосфатазу, вследствие чего снижается активность гомолога белка Ras (Rheb) и ингибируются сигнальные пути мишени для рапамицина млекопитающих (mTOR). Сигнальные пути mTOR регулируют процессы биосинтеза белков и жиров, а также цикл развития клеток, зависящих от факторов роста.
В нормальных условиях гамартин и туберин активируются в процессе биосинтеза, опосредованного комплексом mTORC1, включающим mTOR, субъединицу mTOR – raptor (белок, выполняющий регуляторные функции mTOR), mLST8 и PRAS40 (богатый пролином Akt субстрат 40). Следствием мутации генов TSC1 или TSC2 является избыточная активация сигнального пути mTOR. Нисходящий киназный сигнальный каскад, который запускается в результате мутации данных генов, становится причиной аномалий развития различных клеток, в том числе нарушения клеточного цикла, транскрипции, трансляции и метаболического контроля (рис.).
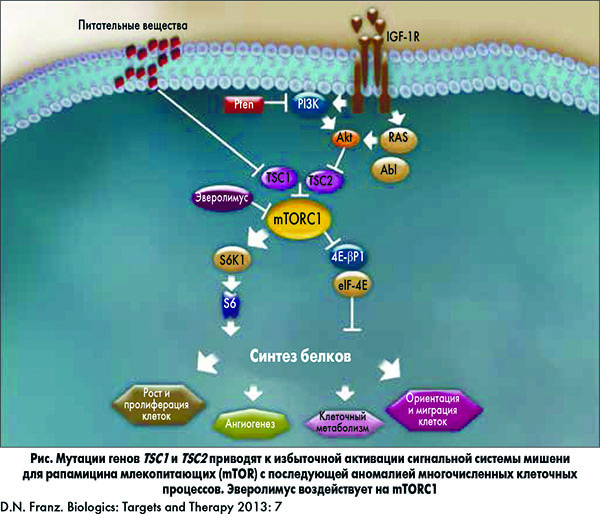
Прогресс в изучении молекулярных основ патофизиологического механизма ТС и выявление критической роли избыточной активации mTOR проложили путь к развитию новых терапевтических стратегий, основанных на применении ингибиторов mTOR.
Выделяют ТС 1 типа, обусловленный мутацией гена TSC1, и ТС 2 типа, обусловленный мутацией гена TSC2. Считается, что мутации в гене TSC2 ассоциированы с более тяжелыми клиническими вариантами заболевания, у пациентов с мутацией TSC1 заболевание протекает легче. Внешние факторы риска для развития ТС отсутствуют.
Диагностика ТС
Диагностика основана на сочетании клинических симптомов и данных лабораторных исследований, но ключевое значение при постановке диагноза имеет комплекс клинических данных. Фенотип пациента с ТС зависит от количества, локализации и размера гамартом. При постановке диагноза также важен возраст пациента, поскольку разные проявления заболевания манифестируют в разные возрастные периоды. Многообразие клинических проявлений, вариабельность фенотипа и зависимость клинических вариантов манифестации заболевания от возраста пациента существенно затрудняют диагностику. Поэтому при постановке диагноза ТС требуется подробный сбор анамнеза, консультации смежных специалистов, а также осмотр родственников пациента. Генетические исследования являются дополнительным методом диагностики и не всегда доступны для широкого применения в клинической практике из-за высокой стоимости.
Для упрощения диагностики ТС были предложены алгоритмы, представленные в Международных рекомендациях (TSC Clinical Consensus Conference 2012). С помощью первичных и вторичных признаков можно с высокой долей вероятности диагностировать ТС.
Диагноз ТС не вызывает сомнений при наличии у пациента двух первичных признаков или одного первичного и двух вторичных.
Первичные (большие) признаки:
• ангиофибромы лица (не менее трех) или фиброзные бляшки на лбу;
• гипопигментные пятна (в количестве не менее трех и не менее 5 мм в диаметре);
• нетравматические околоногтевые фибромы (не менее двух);
• участок «шагреневой кожи»;
• множественные гамартомы сетчатки;
• корковые дисплазии (не менее трех): корковые туберы и миграционные тракты в белом веществе головного мозга;
• субэпендимальные узлы (не менее двух);
• субэпендимальная гигантоклеточная астроцитома;
• рабдомиомы сердца множественные или одиночные;
• лимфангиолейомиоматоз легких;
• множественные АМЛ почек (не менее двух).
Что касается двух последних критериев, то само по себе наличие ЛАМ легких, АМЛ почек или их сочетание может быть не связано с ТС. Для подтверждения диагноза необходимо выявление и других признаков.
Вторичные (малые) признаки:
• многочисленные углубления в эмали зубов (не менее трех);
• фибромы в полости рта (не менее двух);
• гамартомы внутренних органов;
• ахроматический участок сетчатой оболочки глаза;
• пятна «конфетти» на коже;
• множественные кисты почек.
Возможный диагноз ставится на основании наличия одного первичного признака или двух (и более) вторичных. В настоящее время разработано молекулярно-генетическое тестирование. Подтвержденная патогенная мутация TSC1 или TSC2 является дополнительным критерием, достаточным для постановки диагноза ТС в спорных случаях при неполноте клинических данных. В оптимальных условиях генетическое тестирование выявляет мутации у 75-80% пациентов, страдающих ТС. Поэтому отрицательный генетический результат теста не исключает диагноза ТС. Молекулярно-генетическое тестирование полезно в неопределенных или сомнительных случаях, для пренатальной диагностики, а также при скрининге членов семьи и родственников пациента.
Клинические варианты ТС
Поражение ЦНС
Чаще всего поражение ЦНС манифестирует у новорожденных. Приступы судорог, аутизм и задержки физического развития – первые симптомы, требующие исключения ТС. Часто формируются резистентные к терапии варианты эпилепсии. Осложнения, вызванные поражением ЦНС, являются наиболее распространенными причинами смертности при ТС. Это обусловлено главным образом резистентной эпилепсией, эпилептическим статусом, а также формированием гидроцефалии при СЭГА.
Туберы встречаются у 95-100% больных ТС, представляют собой участки фокальной корковой дисплазии со сниженным числом ГАМК-эргических нейронов и характеризуются потерей классической 6-слойной цитоархитектуры коры мозга. Туберы бывают единичные или множественные, размером от нескольких миллиметров до нескольких сантиметров, и располагаются над единичной или несколькими прилегающими бороздами. В 54% случаев туберы кальцифицированы. Существует топографическая связь между наличием фокуса на электроэнцефалограмме и тубером, выявленным при исследовании методом магнитно-резонансной томографии. Приступы могут исходить из окружающей тубер нормальной коры, которая является зоной «перевозбужденных» нейронов. В некоторых случаях тубер может быть «немым», то есть неэпилептогенным.
СЭУ встречаются у 95-98% больных ТС. Они множественные, размером 2-10 мм, локализуются в стенках боковых желудочков, частично или полностью кальцифицированы и бессимптомные. Существует прямая зависимость между тяжестью эпилепсии при ТС и числом корковых туберов и СЭУ. Число туберов (8 и более) является фактором риска манифестации эпилепсии в первые два года жизни ребенка.
Поражение белого вещества головного мозга, или так называемые радиальные миграционные тракты встречаются у 30-95% больных ТС. Они представляют собой группы гетеротопических кластерных клеток, которые соединяют эпендиму стенок желудочков и туберы, соответствуют миграционным путям спонгиобластов во время эмбриогенеза, являются эпилептогенными зонами.
СЭГА встречаются с частотой 5-20% и представляют собой доброкачественную опухоль, которая наблюдается у пациентов с ТС. СЭГА – медленно растущая глионейрональная опухоль, которая не способна к спонтанной регрессии, располагается преимущественно у отверстия Монро (редко – в задних отделах тела боковых желудочков, у височных рогов, в 3 и 4 желудочках) и зачастую имеет >5 мм в диаметре.
Чаще всего СЭГА наблюдаются при мутации TSC2, реже при TSC1. Обычно они образуются у детей в возрасте 4-10 лет, но иногда выявляются антенатально или в период новорожденности. Трансформация СЭУ в СЭГА чаще всего происходит в первые два десятилетия жизни, пик трансформации приходится на пубертатный период. СЕГА обычно имеют круглую или овальную форму и в большинстве случаев не инфильтрируют вещество мозга. Данная опухоль хорошо выявляется с помощью компьютерной томографии с контрастированием.
При увеличении диаметра до ≥10 мм опухоли могут нарушать циркуляцию спинномозговой жидкости и приводить к прогрессирующему увеличению размеров боковых желудочков и развитию тяжелой гидроцефалии.
Симптоматическая эпилепсия наблюдается у 92% больных ТС в течение жизни. У 63% больных эпилептические приступы появляются на первом году жизни. Инфантильные спазмы (относятся к «катастрофическим формам» эпилепсии, приводят к инвалидизации пациентов) регистрируются у 38% пациентов с ТС, при этом пик дебюта заболевания приходится на 3-5 мес жизни. Фокальные приступы предшествуют, сопутствуют инфантильным спазмам и/или следуют за ними. Эпилептические приступы резистентны к противоэпилептической терапии у трети больных ТС. Эпилепсия приводит к нарушению познавательных и социальных функций, интеллекта, слуховой и зрительной памяти, внимания, к трудностями обучения (проблемы с чтением, письмом, арифметическими действиями), а также к жестким социальным ограничениям.
Нарушение обучения встречается у 50% пациентов с ТС. Чем раньше дебютирует ТС, захватывая период от первых месяцев жизни до 1,5 года, тем более выраженно снижается умственное развитие. Нарушения поведения – аутизм (возникает после инфантильных спазмов у 45% пациентов), агрессия и/или аутоагрессия – наблюдаются у 13% больных ТС, также для них характерна гиперактивность. Со временем развивается эмоциональное потускнение наряду с безразличием к родным. Периодически меняется эмоциональное состояние: сниженное настроение обычно сочетается с раздражительностью, агрессивностью; значительно реже наблюдаются дисфория с дурашливостью, недовольством. Также при ТС может отмечаться нарушение сна в виде проблем с засыпанием, частых и ранних пробуждений, сомнамбулизма.
Поражение кожи
Для ТС характерны поражения кожи, которые проявляются в виде гипопигментных пятен, ангиофибром лица, участков «шагреневой кожи», околоногтевых фибром, фиброзных бляшек, а также белых прядей волос.
Гипопигментные пятна – одно из наиболее частых кожных проявлений ТС – встречаются в 90% случаев. Они нередко обнаруживаются с рождения и служат одним из первых манифестных признаков заболевания. С возрастом наблюдается тенденция к увеличению их числа. Гипопигментные пятна при ТС преимущественно локализуются на туловище и ягодицах, реже на лице. Характерной особенностью является асимметричность их расположения. Отмечена вариабельность числа, размера и формы этих пятен. Число гипопигментных пятен варьирует от 3-4 до ≥100. Чаще всего встречаются полигональные пятна размером 0,5-2 см, напоминающие отпечатки большого пальца руки. Наиболее характерная форма пятен – овальная, похожая на лист ясеня, размер их варьирует от 1 до 12 см. Реже встречается большое количество мелких пятен размером 1-3 мм, которые группируются и принимают вид рассыпанного конфетти.
Белые пряди волос, ресниц и бровей (полиозис), как и гипопигментные пятна, представляют собой характерный признак ТС. Наряду с гипопигментными пятнами при ТС в 16% случаев встречаются пигментные пятна цвета кофе с молоком, что не превышает средних популяционных значений. При ТС эти пятна обычно единичные, овальные или округлые, плоские, размером 1-5 см.
Ангиофибромы лица встречаются в 47-90% случаев у детей >5 лет и развиваются, как правило, после 2-5 лет. Внешне они представляют собой папулы 1-4 мм в диаметре с гладкой поверхностью розового или красного цвета. Ангиофибромы располагаются на лице симметрично с двух сторон – на щеках и носу, на подбородке по типу «крыльев бабочки». Иногда ангиофибромы образуют сливные участки.
Участок «шагреневой кожи» представляет собой соединительнотканный невус, служит облигатным признаком ТС и встречается у 50% больных. В большинстве случаев такие участки появляются на втором десятилетии жизни, располагаются преимущественно в пояснично-крестцовой области, имеют плотную консистенцию и желтовато-коричневый или розовый цвет, умеренно выступают над поверхностью окружающей кожи.
Фиброзные бляшки обычно бежевого цвета, шероховатые на ощупь и несколько выступают над окружающей кожей. Они часто образуются уже на первом году жизни и являются одним из первых клинических симптомов заболевания. Чаще всего фиброзные бляшки локализуются на лбу, но иногда встречаются на волосистой части головы. Размер и число бляшек могут варьировать.
Околоногтевые фибромы (опухоли Коэнена) являются облигатным признаком ТС. Они представляют собой тусклые красные или цвета сырого мяса папулы/узлы, растущие от ногтевого ложа или вокруг ногтевой пластинки. В большинстве случаев околоногтевые фибромы появляются на втором десятилетии жизни и склонны к прогрессивному росту и возобновлению даже после их удаления.
Мягкие фибромы встречаются у 30% больных. Это множественные или единичные мягкие образования на ножках, имеющие мешотчатую форму и растущие на шее, туловище и конечностях. Другой вариант мягких фибром – множественные, несколько приподнятые над поверхностью кожи (и такого же цвета) мелкие образования размером меньше булавочной головки, располагающиеся на туловище и шее и напоминающие гусиную кожу.
Поражение сердца
Рабдомиомы относятся к гамартомам – опухолевидным узловым образованиям, представляющим собой тканевую аномалию развития, возникающую в связи с неправильным формированием эмбриональных тканевых комплексов при ТС. Рабдомиомы – один из вариантов манифестации заболевания с поражением сердца.
Клинические симптомы рабдомиом у детей различаются в зависимости от возраста, относительных размеров и локализации опухоли.
Поражение органа зрения
У 50% пациентов с ТС диагностируются гамартомы сетчатки. Встречаются как одно-, так и двусторонние гамартомы. Билатеральные поражения отмечаются примерно у половины пациентов. Обнаружить гамартомы у детей с ТС можно уже в первые дни жизни. Редко гамартомы сетчатки сопровождаются развивитием осложнения, сопровождающиеся снижением зрения: субретинальная экссудация, макулярный отек, кровоизлияния в стекловидное тело, неоваскулярная глаукома.
Патология почек
Как правило, поражение почек при ТС носит прогрессирующий характер, приводя к формированию хронической почечной недостаточности. Она занимает второе место в причинах смертности при ТС после патологии нервной системы. Типичным поражением почек являются АМЛ и кисты. К редким видам патологии относятся онкоцитома и почечноклеточный рак, а также неопухолевые заболевания: нефролитиаз, фокально-сегментарный гломерулосклероз, сосудистые дисплазии, мезангиокапиллярный гломерулонефрит.
АМЛ – наиболее частая патология почек при ТС, которая выявляется у 75% детей и 80% взрослых при ультразвуковом исследовании. АМЛ состоят из количественно вариабельных эндотелиальных гладкомышечных клеток и жировой ткани. Это доброкачественная опухоль, редко выявляемая при рождении и обычно диагностируемая в возрасте 5-10 лет. Обычно АМЛ развивается в обеих почках и имеет тенденцию к росту в юношеском возрасте. Размеры новообразования могут достигать 30 см и редко прорастают в окружающие ткани. У пациентов с ТС АМЛ почек часто сочетаются с ЛАМ легких (прогрессирующим кистозным поражением легких, встречающимся преимущественно у женщин).
Кисты почек при ТС могут развиваться в любом отделе нефрона и бывают как единичными, так и множественными. У большинства пациентов кисты невелики по размеру и не представляют значительного риска для здоровья. Однако в 2-5% случаев присутствуют изменения, аналогичные таковым при аутосомно-доминантной поликистозной болезни. Это связано с делецией генов ТSC2 и PKD1, расположенных на коротком плече 16 хромосомы и непосредственно примыкающих друг к другу. При этом патология почек проявляет себя в более раннем возрасте, может быть диагностирована пренатально при УЗИ плода и протекает более тяжело, чем при аутосомно-доминантной поликистозной болезни, связанной только с мутацией гена PKD1. Кисты интенсивно растут, достигая ≥5 см в диаметре и приводя к артериальной гипертензии и хронической почечной недостаточности уже к подростковому возрасту.
Поражение желудочно-кишечного тракта, эндокринной и костной систем
Изменения в органах желудочно-кишечного тракта при ТС разнообразны, встречаются относительно часто и проявляются патологией ротовой полости, печени, селезенки, поджелудочной железы и прямой кишки. Наиболее типичными нарушениями, выявляемыми при исследовании ротовой полости, являются узловые опухоли, фибромы или папилломы. Они локализуются в основном на переднем крае десен, преимущественно на верхней челюсти, но также встречаются на губах, слизистой оболочке щек, спинке языка и небе.
Дефекты эмали зубов отмечаются практически у всех больных ТС. Одним из наиболее типичных нарушений является дефект эмали зубов в виде углублений, число которых варьирует от 1 до 11 на каждом зубе (в среднем по 3 углубления на зуб). Преимущественные зоны локализации данных дефектов не определяются.
При ТС у 25% больных в печени появляются одиночные или множественные АМЛ и липомы. В поджелудочной железе АМЛ выявляются реже.
Изменения в кишечнике при ТС проявляются в основном ректальными полипами, которые встречаются, по данным разных авторов, в 50-78% случаев. Как правило, ректальные полипы выявляются у больных >20 лет. Полипы чаще многочисленные, розового цвета, размером 2-4 мм. Локализуются в прямой кишке преимущественно у аноректального соединения. Полипы при ТС обычно прогностически благоприятны.
Дисфункция желез внутренней секреции возникает вследствие роста опухолей. Наиболее часто при ТС обнаруживается патология надпочечников, проявляющаяся преимущественно в виде АМЛ (у 25% больных). Реже встречаются одиночные или множественные аденомы коры надпочечников. Нередко дисфункция желез внутренней секреции при ТС носит плюригландулярный характер.
Патология костной системы при ТС проявляется участками склероза костей свода черепа, чаще в лобной или теменной костях. Они обыкновенно выявляются после десятилетнего возраста и представляют собой образования округлой формы размером 0,2‑2 см. Схожие образования развиваются в телах позвонков и тазовых костях с тенденцией к расположению у крестцово-подвздошного сочленения. Участки склероза, расположенные диффузно по периферии тазовых костей, имеют плохо очерченные границы, вследствие чего могут быть ошибочно приняты за метастазы остеобластомы.
Связанные с ТС патологические образования в костях конечностей отличаются от образований в других частях скелета, представлены участками кистозного разрежения и часто сочетаются с периостальной (надкостничной) дополнительной новой костью. Кистозные образования встречаются преимущественно на руках и выявляются на фалангах пальцев в раннем детском возрасте. Периостальные дополнительные кости, наоборот, появляются в зрелом возрасте чаще на плюсневых, чем на пястных костях. Они плотные, солидные и имеют специфический волнистый контур. Поражение ребер и длинных трубчатых костей для ТС нехарактерно.
Поражение легких
Приблизительно в 30-40% случаев, при ТС развивается ЛАМ, встречаясь преимущественно у женщин репродуктивного возраста; описаны единичные случаи у мужчин и детей. После поражения ЦНС и почек ЛАМ является третьей причиной смерти при ТС. В зависимости от степени выраженности и распространения заболевания макроскопические изменения варьируют от единичных кист до диффузных кистозных изменений с обеих сторон от верхушек до оснований легких. Кистозные изменения в легких могут сопровождаться изменениями в подмышечных лимфатических узлах с формированием кист с жидкостным содержимым. Гистопатологически ЛАМ характеризуется кистами и пролиферацией незрелых гладкомышечных клеток.
Таким образом, ТС представляет собой генетическое мультисистемное заболевание, проявляющееся нарушением клеточной дифференцировки, пролиферации и миграции клеток на ранних стадиях развития, приводит к образованию гамартом практически во всех системах органов человеческого тела и обусловливает разнообразие клинического проявления заболевания. Вариабельность клинического фенотипа пациента требует мультидисциплинарного подхода к диагностике ТС. Сегодня разработаны современные методы таргетной терапии пациентов с некоторыми формами ТС, базирующиеся на использовании специфических ингибиторов, блокирующих mTOR сигнальный путь, активация которого лежит в основе клинико-патогенетических изменений, сопровождающих данное заболевание.
Список литературы находится в редакции.
Подготовила Катерина Котенко
СТАТТІ ЗА ТЕМОЮ Онкологія та гематологія
Гостра лімфобластна лейкемія (ГЛЛ) є найпоширенішим онкогематологічним захворюванням у дітей і складає значну частку серед лейкемій у дорослих. Незважаючи на значні успіхи в лікуванні ГЛЛ у дітей, де рівень виліковності сягає 90%, результати терапії у дорослих залишаються незадовільними. У рамках науково-практичної конференції з міжнародною участю «Діагностика та лікування гематологічних захворювань: підведення підсумків 2023 року» (15-16 грудня 2023 року) проведено секцію, присвячену ГЛЛ....
Хронічна лімфоцитарна лейкемія (ХЛЛ) залишається актуальною проблемою сучасної онкогематології. Незважаючи на певні досягнення в терапії, ХЛЛ є невиліковним захворюванням. Стандартна хіміотерапія не забезпечує стійкої відповіді, а трансплантація гемопоетичних стовбурових клітин можлива лише для окремої когорти пацієнтів. Тому пошук нових підходів до терапії ХЛЛ, зокрема таргетної, є нагальним завданням. ...
Гепатоцелюлярна карцинома (ГЦК) – злоякісне новоутворення в печінці, що розвивається з гепатоцитів. Рання діагностика і початок лікування пацієнтів із ГЦК запобігає виникненню тяжких ускладнень і покращує якість життя пацієнтів. Медична допомога пацієнтам із ГЦК потребує міждисциплінарної співпраці та інтегрованого ведення хворих мультидисциплінарною командою фахівців, яка займається або спеціалізується на злоякісних новоутвореннях печінки. Саме цьому сприятимуть положення Стандарту медичної допомоги «Гепатоцелюлярна карцинома»....
Традиційно січень є місяцем обізнаності про рак шийки матки (РШМ) – однієї з найпоширеніших патологій у структурі онкогінекологічних захворювань. Протягом цього місяця світ забарвлюється в палітру бірюзового та білого з метою привернення уваги громадськості до проблеми РШМ. ...