



Довготривалі ефективність і безпечність таліглюцерази альфа у дітей із хворобою Гоше
Хвороба Гоше (ХГ) – лізосомна хвороба накопичення, що розвивається внаслідок мутацій гена GBA1, який кодує синтез кислої β-глюкозидази (глюкозидцереброзидази). Недостатня активність цього ферменту призводить до накопичення глюкоцереброзиду (сфінголіпіду) в лізосомах різних клітин і тканин з подальшим розвитком вісцеральних, кісткових і неврологічних симптомів. Хвороба отримала назву за прізвищем французького лікаря Філіпа Гоше, який уперше описав її 1882 року в пацієнта зі спленомагалією без лейкозу.
Поширеність ХГ у загальній популяції становить від 1 на 40 000 до 1 на 60 000 народжень, проте може досягати 1 на 800 народжень в євреїв ашкеназі.
ХГ має три основні клінічні типи. Найбільш поширеною (90% пацієнтів у США та Європі) є ХГ I типу, яка проявляється гепатоспленомегалією, анемією, тромбоцитопенією, ураженням кісток і кісткового мозку за відсутності первинного ураження центральної нервової системи (таке ураження властиве ХГ II і III типу). У пацієнтів, у яких ХГ I типу діагностовано в ранньому дитинстві, захворювання має тенденцію до тяжчого перебігу та швидшого прогресування. За даними дослідження Weinberg і співавт. (2008), які проаналізували дані 2876 пацієнтів із ХГ I типу (загалом 13 509 пацієнто-років спостереження), очікувана тривалість життя таких хворих у середньому на 9 років менша порівняно із загальною популяцією.
Слід зазначити, що 90% пацієнтів отримували замісну ферментну терапію (ЗФТ). ЗФТ із періодичними інфузіями рекомбінантної кислої β-глюкозидази ефективно зменшує вісцеральні прояви ХГ і є першою лінією лікування пацієнтів з ХГ I і III типу.
До рідкісних, або орфанних, належать хвороби, які уражають малу кількість людей. Більшість таких захворювань є спадковими і наявні протягом усього життя людини. Багато рідкісних хвороб маніфестують у ранньому віці, і близько 30% дітей із ними помирають до свого 5-річчя. Найбільш рідкісним генетичним захворюванням (на сьогоднішній день діагностоване лише в 1 пацієнта) є дефіцит рибозо‑5-фосфатізомерази. Якогось порогового критерію «рідкісності» не існує, оскільки та сама хвороба може вважатися рідкісною в одній частині світу (або в окремій популяції) і зустрічатись досить часто в іншій.
Перші спроби лікувати пацієнтів із ХГ внутрішньовенним введенням ферментних препаратів виявилися невдалими: не забезпечували достатнього терапевтичного ефекту через те, що екзогенний немодифікований людський фермент, отриманий із плаценти, не досягав своїх внутрішньоклітинних мішеней. До того ж можливості вироблення такого ферменту у достатніх кількостях були дуже обмеженими. Виробництво сучасних препаратів для ЗФТ стало можливим завдяки здатності продукувати людський фермент у великій кількості за допомогою рекомбінантних методів і відкриттю макрофагальних манозних рецепторів (MMR).
Сьогодні в лікуванні ХГ I і III типу використовують три препарати рекомбінантної кислої β-глюкозидази. Іміглюцераза та велаглюцераза альфа виробляються лініями клітин ссавців (яєчників китайського хом’яка та фібросаркоми людини відповідно). Для виготовлення таліглюцерази альфа (Елелісо®, Protalix/Pfizer) використовують клітини генетично модифікованої моркви. Ця інноваційна технологія має низку переваг порівняно із застосуванням клітин ссавців, таких як повна ізольованість і стерильність системи виробництва, відсутність ризику контамінації людськими патогенами, кращі можливості післятрансляційної модифікації та нижча вартість виробництва. Ефективність і безпечність таліглюцерази альфа вивчали у низці клінічних досліджень III і IV фази.
Парадокс рідкісних хвороб полягає в тому, що кожне захворювання хоча і зустрічається рідко, та в сукупності всі пацієнти з рідкісними хворобами становлять величезну популяцію. Наприклад, у країнах Євросоюзу сьогодні зареєстровано близько 30 млн таких пацієнтів, в усьому світі – близько 300 млн. Загалом на сьогоднішній день відомо близько 7 тис. рідкісних захворювань (вроджених, гематологічних, онкологічних, аутоімунних тощо), які виявляють у 6-8% населення планети.
У дослідженні PB‑06-006 вивчали довготривалі ефективність і безпечність таліглюцерази альфа в пацієнтів із ХГ віком від 2 до 18 років, які завершили участь у дослідженнях PB‑06-005 або PB‑06-002.
PB‑06-005 було подвійним сліпим дослідженням, у якому раніше не лікованих пацієнтів з ХГ рандомізували для лікування таліглюцеразою альфа в дозі 30 або 60 од./кг упродовж 12 міс. PB‑06-002 було відкритим дослідженням за участі дітей і дорослих зі стабільним перебігом ХГ, які протягом ≥2 років отримували стабільну підтримувальну дозу іміглюцерази; цих хворих переводили на лікування таліглюцеразою альфа і спостерігали впродовж 9 міс.
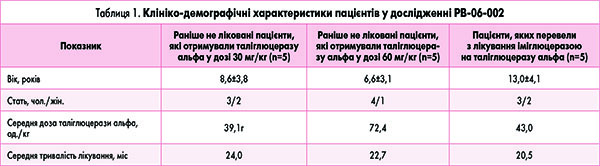
У дослідженні PB‑06-006 пацієнти продовжували отримувати інфузії таліглюцерази альфа 1 раз на 2 тижні у тій самій дозі протягом додаткових 24 міс. Отже, загальна тривалість терапії для пацієнтів з досліджень PB‑06-005 (n=10) і РВ‑06-002 (n=5) становила 36 і 33 міс відповідно (табл. 1). Два пацієнти мали ХГ III типу, інші 13 – ХГ І типу.
Зміна параметрів ефективності лікування порівняно з початковими показниками представлена в таблиці 2. У раніше не лікованих пацієнтів об’єм селезінки значно зменшився після 36 міс терапії: в середньому з 23,9 до 9,0 і з 29,4 до 6,6 MN у хворих, які отримували таліглюцеразу альфа у дозі 30 і 60 од./кг відповідно. У пацієнтів, які були переведені на лікування таліглюцеразою альфа, цей показник покращувався або залишався стабільним упродовж 33 міс лікування. Подібна позитивна динаміка спостерігалась і щодо об’єму печінки. Лікування таліглюцеразою альфа в обох дозах також значно покращувало рівні гемоглобіну і тромбоцитів. У жодного з 7 пацієнтів, у яких на момент включення зафіксовано анемію, наприкінці періоду спостереження цей симптом не спостерігали. У хворих, що були переведені на терапію таліглюцеразою альфа з іміглюцерази, концентрації гемоглобіну та тромбоцитів залишалися стабільними або покращувались.

Концентрація ліганду 18 хемокіну (С-С-мотиву; CCL18) і активність хітотріозидази значно покращувалися у раніше не лікованих пацієнтів, причому найбільш значні зміни відбувалися після 12 міс терапії таліглюцеразою альфа. У хворих, які отримували таліглюцеразу альфа замість іміглюцерази, ці сурогатні маркери ефективності лікування ХГ також покращувалися.
Орфанні препарати – лікарські засоби, призначені для діагностики, профілактики чи лікування рідкісних захворювань або станів, що становлять серйозну загрозу для здоров’я або життя пацієнта. Препарати називаються орфанними, або «сирітськими», тому що в умовах ринкової економіки фармацевтична промисловість не зацікавлена в розробленні та продажу ліків, призначених для невеликої кількості хворих. Це пояснюється тим, що очікувані надходження від продажу таких лікарських засобів не покривають надзвичайно високих витрат фармацевтичних компаній, пов’язаних із їх виведенням на ринок. Саме тому уряди й організації, що об’єднують пацієнтів із рідкісними захворюваннями (такі як National Gaucher Foundation), шукають можливості для економічного стимулювання фармацевтичних компаній, які займаються розробленням і виробництвом лікарських засобів для лікування рідкісних захворювань.
В усіх пацієнтів, включених у дослідження, протягом періоду спостереження реєстрували нормальну динаміку росту і кісткового віку.
Якість життя визначали у 9 раніше не лікованих пацієнтів за допомогою опитувальника CHQ-PF28. Наприкінці періоду спостереження загальний стан здоров’я дитини як «дуже добрий» оцінили 7 з 9 батьків (до початку лікування – лише 1 з 9).
Лікування таліглюцеразою альфа добре переносилось. Всі небажані реакції були легкими або помірними. Найчастіше (≥3 з 15 пацієнтів) спостерігали кашель (n=4), головний біль (n=4), інфекцію верхніх дихальних шляхів (n=4), біль у животі (n=3), лихоманку Денге (n=3), діарею (n=3), лімфатичний набряк (n=3), назофарингіт (n=3) і біль у кінцівках (n=3). Лише 1 небажана реакція (незначний біль у місці ін’єкції) була розцінена як потенційно пов’язана з досліджуваним препаратом. У жодного пацієнта не було потреби змінювати дозу або відміняти препарат через побічні реакції.
Під час дослідження PB‑06-005 в 1 пацієнта, який отримував препарат у дозі 30 од./кг, визначено IgG-антитіла проти таліглюцерази альфа. Протягом усього періоду дослідження PB‑06-006 хворий залишався позитивним за цими антитілами; незважаючи на це, після 36 міс лікування таліглюцеразою альфа в нього виявлено значне покращення об’єму печінки й селезінки, рівнів гемоглобіну й тромбоцитів, активності хітотріозидази і концентрації ССL18. У жодного пацієнта, що були переведені з іміглюцерази на таліглюцеразу альфа, антитіла проти останньої не визначали.
Отже, дослідження PB‑06-006 засвідчило, що у дітей з ХГ лікування таліглюцеразою альфа в дозі 30 або 60 од./кг протягом 36 міс забезпечувало прогресивне покращення вісцеральних і гематологічних параметрів, а також біомаркерів хвороби. У дітей, переведених на лікування таліглюцеразою альфа з іміглюцерази, впродовж 33 міс визначали стабілізацію або покращення цих показників. Зменшення вісцеральних і гематологічних проявів у дітей із ХГ було подібним до такого, що спостерігали в дослідженнях іміглюцерази й велаглюцерази альфа за участі дітей і дорослих із ХГ. Терапія таліглюцеразою альфа добре переносилась, будь-яких нових сигналів щодо безпечності препарату не реєстрували.
Докладніше про дослідження PB-06-006:
Zimran A., Duran G., Mehta A. et al. Long-term efficacy and safety results of taliglucerase alfa up to 36 months in adult treatment-naive patients with Gaucher disease. Am J Hematol. 2016 Jul;91(7):656-60.
Підготував Олексій Терещенко
Надруковано за підтримки компанії Файзер.
WUKELE0317004
СТАТТІ ЗА ТЕМОЮ Педіатрія
Вроджена дисфункція кори надниркових залоз (ВДКНЗ) – це захворювання з автосомно-рецесивним типом успадкування, в основі якого лежить дефект чи дефіцит ферментів або транспортних білків, що беруть участь у біосинтезі кортизолу. Рання діагностика і початок лікування пацієнтів з ВДКНЗ сприяє покращенню показників виживаності та якості життя пацієнтів....
Алергічний риніт (АР) є поширеним запальним захворюванням верхніх дихальних шляхів (ВДШ), особливо серед педіатричних пацієнтів. Ця патологія може знижувати якість життя, погіршувати сон та щоденну продуктивність. Метою наведеного огляду є надання оновленої інформації щодо епідеміології АР та його діагностики, з урахуванням зв’язку з бронхіальною астмою (БА). ...
Американська академія педіатрії (AAP) оновила рекомендації щодо контролю грипу серед дитячого населення під час сезону 2023-2024 рр. Згідно з оновленим керівництвом, для профілактики та лікування грипу в дітей необхідно проводити планову вакцинацію з 6-місячного віку, а також своєчасно застосовувати противірусні препарати за наявності показань. ...
Поширеність і вплив алергічних захворювань часто недооцінюють [1]. Ключовим фактором алергічної відповіді є імуноглобулін (Ig) Е, присутній на поверхні тучних клітин і базофілів. Взаємодія алергену з IgЕ та його рецепторним комплексом призводить до активації цих клітин і вивільнення речовин, у тому числі гістаміну, які викликають симптоми алергії [2]. Враховуючи ключову роль гістаміну в розвитку алергічних реакцій, при багатьох алергічних станах, включаючи алергічний риніт і кропив’янку, пацієнту призначають антигістамінні препарати [3, 4]....