



Ураження печінки та селезінки при лізосомальних хворобах накопичення у дорослих
 Ураження печінки та селезінки трапляються у клінічній практиці лікаря дуже часто, серед них найбільш поширені – алкогольна та неалкогольна жирова хвороба печінки (АЖХП та НАЖХП), вірусні гепатити, ураження печінки, пов’язані з прийомом ліків. Що стосується ураження селезінки, це здебільшого пацієнти з портальною гіпертензією (ПГ) і гематологічними захворюваннями. За наявності у пацієнта ознак ураження печінки та/або селезінки більшість лікарів-практиків взагалі не проводять діагностичних досліджень для виявлення рідкісних лізосомальних хвороб накопичення (ЛХН), які у представників певних етнічних груп є не такими й рідкісними. Багато лікарів, на жаль, мають хибне уявлення про те, що ЛХН як спадкові захворювання виявляють тільки в дитячому віці. Більшість із них дійсно дебютують із моменту народження і дуже швидко призводять до смерті дитини у ранньому віці, однак є кілька захворювань, симптоми яких з’являються в підлітковому або дорослому віці [1]. Тривалість життя таких пацієнтів не відрізняється від середньостатистичної [9]. У статті розглянуто основні та найбільш поширені ЛХН, які супроводжуються ураженнями печінки та/або селезінки у дорослих та можуть трапитися у практиці терапевта, гастроентеролога та лікарів інших спеціальностей.
Ураження печінки та селезінки трапляються у клінічній практиці лікаря дуже часто, серед них найбільш поширені – алкогольна та неалкогольна жирова хвороба печінки (АЖХП та НАЖХП), вірусні гепатити, ураження печінки, пов’язані з прийомом ліків. Що стосується ураження селезінки, це здебільшого пацієнти з портальною гіпертензією (ПГ) і гематологічними захворюваннями. За наявності у пацієнта ознак ураження печінки та/або селезінки більшість лікарів-практиків взагалі не проводять діагностичних досліджень для виявлення рідкісних лізосомальних хвороб накопичення (ЛХН), які у представників певних етнічних груп є не такими й рідкісними. Багато лікарів, на жаль, мають хибне уявлення про те, що ЛХН як спадкові захворювання виявляють тільки в дитячому віці. Більшість із них дійсно дебютують із моменту народження і дуже швидко призводять до смерті дитини у ранньому віці, однак є кілька захворювань, симптоми яких з’являються в підлітковому або дорослому віці [1]. Тривалість життя таких пацієнтів не відрізняється від середньостатистичної [9]. У статті розглянуто основні та найбільш поширені ЛХН, які супроводжуються ураженнями печінки та/або селезінки у дорослих та можуть трапитися у практиці терапевта, гастроентеролога та лікарів інших спеціальностей.
Лізосомальні хвороби накопичення (англ. Lysosomal Storage Diseases) – загальна назва групи рідкісних спадкових захворювань, причиною яких є порушення функції внутрішньоклітинних органел лізосом [1, 9]. Ці одномембранні органели здійснюють внутрішньоклітинне розщеплення глікогену, глікозаміногліканів, глікопротеїнів тощо [1]. Причина розвитку ЛХН – генетично зумовлений дефіцит ферментів лізосом, що призводить до накопичення макромолекул, які є субстратом цих ферментів у різних органах і тканинах організму.
До цієї групи належать мукополісахаридози, муколіпідози, глікогенози, хвороби накопичення ліпідів, глікопротеїнів та інших макромолекул [1].
Хвороба Гоше
Одна з найбільш поширених форм спадкових ферментопатій, об’єднаних у групу ЛХН за наявністю дефекту лізосомного ферменту β-D-глюкозидази (глюкоцереброзидази), що відповідає за катаболізм ліпідів [8].
Цю хворобу вперше описав у 1882 р. французький лікар P.C.E. Gaucher. Він ідентифікував патогномонічні для цього захворювання клітини-макрофаги, які накопичують ліпіди, пізніше вони отримали назву «клітини Гоше» [14].
Частота хвороби Гоше (ХГ) у загальній популяції 1:40000-1:70000. У популяції євреїв-ашкеназі (вихідців зі Східної Європи) її частота є вищою і сягає 1:450 [16].
ХГ успадковується за аутосомно-рецесивним типом. Наявність двох мутантних алелів гена асоціюється зі значним (приблизно на 30% від нормального рівня) зниженням каталітичної активності глюкоцереброзидази, що призводить до накопичення в лізосомах макрофагів неутилізованих ліпідів та утворення клітин Гоше. Наслідками цього метаболічного дефекту є: хронічна активація макрофагальної системи, аутокринна стимуляція моноцитопоезу та збільшення абсолютної кількості макрофагів, порушення регуляторних функцій макрофагів. Ген глюкоцереброзидази картований на хромосомі 1q21. Сьогодні описано понад 300 різних мутацій, які частково або повністю блокують каталітичну активність ферменту та пов’язані з широким поліморфізмом клінічних симптомів ХГ. Найбільш поширеними мутаціями (складовими близько 60% всіх мутацій гена глюкоцереброзидази) є N370S, L444P та 84GG. Наявність алеля N370S запобігає ураженню центральної нервової системи (ЦНС) [8, 16].
Залежно від особливостей клінічного перебігу виділяють три типи ХГ:
- тип І – ненейронопатичний (найбільш поширений);
- тип ІІ – інфантильний, або гострий нейронопатичний;
- тип ІІІ – підгострий нейронопатичний.
ХГ І типу має хронічний перебіг. Клінічна картина характеризується прогресуючим збільшенням паренхіматозних органів (печінки та селезінки), панцитопенією і патологією трубчастих кісток скелету. Характерними ознаками ХГ І типу є:
- гепатоспленомегалія;
- кісткові болі (кісткові кризи);
- патологічні переломи;
- порушення рухливості в суглобах, що часто зумовлені асептичним некрозом;
- геморагічний синдром;
- затримка фізичного і статевого розвитку;
- астенічний синдром.
Типова і найбільш рання ознака захворювання – спленомегалія. Розміри селезінки при ХГ можуть перевищувати норму в 5-80 разів. При пальпації селезінка має щільну консистенцію. З прогресуванням спленомегалії в селезінці можуть розвиватися інфаркти та фіброзні зміни, часто за відсутності клінічних проявів [1].
Гепатомегалія спостерігається у 80-90% пацієнтів із ХГ I типу. Розміри печінки збільшуються в 2-4 рази, що значно менше порівняно із селезінкою. Від 30 до 50% хворих мають незначне підвищення активності амінотрансфераз сироватки крові, зазвичай не більше ніж удвічі від норми, інші показники функції печінки, як правило, не змінюються. Стандартна біопсія печінки завжди виявляє клітини Гоше (рис. 1), які містяться у вигляді агрегатів у синусоїдах або дифузно розподілені в усіх зонах і часточках у біоптаті [9].
.jpg)
Найбільш поширеним і раннім проявом цитопенічного синдрому є тромбоцитопенія, що супроводжується спонтанним геморагічним синдромом у вигляді підшкірних гематом, кровоточивості слизових оболонок і тривалими кровотечами після малих оперативних втручань. Пізніше розвиваються анемія і нейтропенія [10].
При прогресуванні ХГ може розвинутися ПГ. Фактор ризику, достовірно пов’язаний із розвитком ПГ, – попередня спленектомія [8].
Стандартом сучасної діагностики є біохімічний аналіз активності глюкоцереброзидази в лейкоцитах крові. Діагноз підтверджується при зниженні активності ферменту до рівня 30% і менше від норми. Додатковим характерним біохімічним маркером є значне підвищення активності хітотріозидази в сироватці крові. Верифікувати діагноз можна за допомогою молекулярного аналізу гена глюкоцереброзидази: наявність двох мутантних алелів підтверджує діагноз ХГ [8].
Наявність численних клітин Гоше в пунктаті й трепанобіоптаті кісткового мозку або біоптаті печінки є доказовою ознакою ХГ. Однак при цьому слід враховувати, що поодинокі клітини з аналогічною морфологією (Гоше-подібні) можуть зустрічатися при інших захворюваннях, що супроводжуються підвищеною деструкцією клітин, наприклад при хронічному мієлолейкозі та лімфопроліферативних захворюваннях [8, 9].
Рентгенографія кісток скелета, денситометрія та МРТ – методи, які дозволяють виявити ураження кісток при ХГ.
Лікування ХГ полягає в довічному призначенні замісної ферментної терапії рекомбінантної глюкоцереброзидази (іміглюцерази). Іншим напрямом у лікуванні ХГ є субстратредукційна терапія, першим представником якої є лікарський препарат міглустат, призначений для лікування легкої форми ХГ за умови неможливості проведення замісної ферментної терапії рекомбінантної глюкоцереброзидази [8].
Сучасна тактика ведення пацієнтів із ХГ зазнала істотних змін порівняно з відповідною тактикою кінця минулого століття. Зараз встановлено достовірний зв’язок між спленектомією в анамнезі та подальшим несприятливим перебігом захворювання. Тому спленектомія, яку раніше застосовували, виключена з переліку методів лікування ХГ. Діагностичну спленектомію у хворих зі спленомегалією невстановленого генезу слід проводити тільки після виключення діагнозу ХГ [8].
За наявності доведеного діагнозу ХГ не слід проводити інвазивні діагностичні повторні пункції і трепанобіопсії кісткового мозку для контролю цитопеній та біопсію печінки і/або селезінки з метою підтвердження діагнозу. Протипоказано застосовувати кортикостероїди для лікування цитопенічного синдрому. Необґрунтованим є призначення препаратів заліза хворим із розгорнутою клінічною картиною ХГ через те, що анемія в цьому випадку не пов’язана з дефіцитом заліза [8, 9].
Хвороба Німана-Піка
Це захворювання вперше описав німецький педіатр A. Niemann в 1914 р., а L. Pick в 1927 р. узагальнив результати клініко-патологоанатомічних спостережень кількох хворих та визначив характерні гістологічні критерії, що властиві цьому захворюванню [1, 6].
Виділяють 3 типи хвороби Німана-Піка (ХНП). Для дорослих актуальними є типи В і С. ХНП має аутосомно-рецесивний тип успадкування. Частота ХНП типу А і В приблизно становить 1:100 000 і ХНП типу С – 1:150 000. Захворювання зустрічається у представників різних етнічних груп, однак найчастіше (в 30-50% всіх описаних випадків) у євреїв-ашкеназі (так само, як і ХГ) [12, 13].
Прогресування ХНП типу В пов’язане із мутаціями в гені сфінгомієлін фосфодієстерази I (SMPD-I), який кодує фермент – кислу сфінгомієліназу. У результаті мутацій у гені SMPD-I знижується її активність. При цьому порушується розщеплення сфінгомієліна на фосфохолін і церамід і відбувається його накопичення в клітинах усіх органів і тканин. При ХНП типу B сфінгомієлін накопичується переважно у внутрішніх органах і практично не відкладається у головному мозку [12].
Прогресування ХНП типу С спричиняє порушення структури трансмембранного білка, який бере участь у перенесенні екзогенного холестерину, що пов’язано з мутаціями у гені NPC1 (локус 18q11-q12 хромосоми 18) та у гені NPC2 (локус 14q24 хромосоми 14), які призводять до накопичення неетерифікованого холестерину (ХС) у клітинах [13].
При типі В (вісцеральна форма, хронічна форма без залучення нервової системи) основні клінічні прояви з’являються пізніше, ніж при типі А. Спленомегалія розвивається у віці 2-6 років, пізніше – ураження печінки та легень (пацієнти часто хворіють на інфекції дихальних шляхів). Симптоматика ураження ЦНС відсутня, навпаки, у багатьох пацієнтів відзначено високі інтелектуальні здібності. Тривалість життя пацієнтів не знижена, тобто вони продовжують або, у разі несвоєчасного звернення, починають лікуватися у терапевтів і гастроентерологів [1].
Тип С (підгостра, юнацька форма, хронічна нейропатична форма) найчастіше проявляється у віці 1-2 років і характеризується нейровісцеральними порушеннями. Системні симптоми ХНП типу С включають гепатоспленомегалію і пов’язані з нею симптоми. Легенева інфільтрація пінистими клітинами, як правило, спостерігається тільки у пацієнтів із раннім початком захворювання або з мутаціями в гені NPC2. ХНП типу С може проявлятися захворюваннями печінки в дитячому віці. У пацієнтів із пре- і перинатальною формою ХНП цього типу спостерігається одна або кілька таких ознак: багатоводдя плоду, асцит, неонатальний холестаз, гепатоспленомегалія і/або печінкова недостатність [13].
Гепатоспленомегалія у пацієнтів із початком захворювання в старшому віці є зазвичай безсимптомною і часто клінічно не розпізнається, що при підозрі на наявність ХНП типу С потребує проведення ультразвукового дослідження (УЗД) органів черевної порожнини (ОЧП) [1, 12]. За даними літератури, вона, імовірно, відсутня або має незначну вираженість приблизно у 15% всіх пацієнтів із ХНП типу С і майже у половини пацієнтів із початком захворювання в підлітковому/дорослому віці. Опубліковані дані, ймовірно, занижують показники поширеності гепатоспленомегалії, тому що УЗД ОЧП часто не проводять. За даними УЗД в одній когорті співвідношення пацієнтів зі спленомегалією (з гепатомегалією або без неї) становило до 90%, навіть якщо захворювання розвинулося в дорослому віці. Незважаючи на те що спленомегалія практично завжди спостерігається при ХНП типу С, у дорослих гепатомегалія спостерігається рідше [1, 13].
При патоморфологічному дослідженні макроскопічно визначають збільшення розмірів і щільності печінки, селезінки, лімфатичних вузлів. Поверхня зрізу селезінки має жовтувато-рожевий відтінок, а лімфатичних вузлів і печінки – жовтий. При світловій мікроскопії виявляють у багатьох органах і тканинах клітини з ліпідними включеннями. Цитоплазма клітин виглядає пінистою через численні вакуолі. У результаті досліджень печінки і селезінки виявляють накопичення сфінгомієліна і неетерифікованого ХС [13].
Неврологічні симптоми розвиваються на тлі ураження внутрішніх органів. Спостерігаються м’язова гіпотонія, посилення глибоких сухожильних рефлексів, які змінюються спастичним паралічем, а також інтенційний тремор, помірна атаксія, судоми. У підлітковому або юнацькому віці у пацієнтів із цим типом ХНП часто наявні симптоми психозу, циркулярні розлади, депресія, атипові шизофреноподібні розлади і/або інші психіатричні симптоми, у тому числі синдром дефіциту уваги [1, 13].
У пацієнтів із нейродегенеративними і/або психічними порушеннями наявність ізольованої спленомегалії за відсутності захворювання печінки потребує виключення ХНП типу С [13].
Системні симптоми завжди передують неврологічним. Більше того, вік, коли з’явилися системні симптоми, не пов’язаний із віком, в якому почали проявлятися неврологічні. Слід зауважити, що неврологічні симптоми можуть з’явитися через багато років або навіть десятиліть після появи системних симптомів [13].
Для підтвердження діагнозу визначають активність сфінгомієлінази в культурі фібробластів шкіри і лейкоцитах (при ХНП типу А і В), виявляють накопичення неетерифікованого ХС у культурі фібробластів шкіри (при ХНП типу С), проводять дослідження на наявність генетичних дефектів в 11, 14, 18-й хромосомах [12].
Пункція кісткового мозку у таких хворих виявляє специфічні «пінисті» клітини Німана-Піка (вони мають такий вигляд через накопичення жирів).
Лікування таких пацієнтів проводиться постійно. Основним лікарським засобом є міглустат. Він блокує фермент, що відповідає за синтез глікосфінголіпідів (попередників сфінгомієліна). Дозування залежить від віку і площі тіла хворого. Міглустат запобігає руйнуванню нервових клітин і, таким чином, уповільнює розвиток неврологічних симптомів, приводить до збільшення тривалості життя пацієнтів. Помітний позитивний ефект від застосування препарату відзначається через 6-12 міс його постійного прийому [13].
Окрім цього, проводиться симптоматичне лікування для полегшення страждань хворого із застосуванням таких лікарських засобів: протисудомних, антидепресантів, вальпроатів, протидіарейних, антибіотиків, бронходилататорів, антихолінергічних препаратів [12, 13].
Дефіцит лізосомальної кислої ліпази
Клінічно дефіцит лізосомальної кислої ліпази (ДЛКЛ) може проявлятися в двох формах: у вигляді хвороби Вольмана у дітей у віці до одного року і хвороби накопичення ефірів холестерину (ХНЕХ) в старших вікових групах. Ці захворювання вперше було описано у 1956 р. та у 1963 р. відповідно [2, 4].
Встановлено, що загалом у країнах ЄС мутантний алель E8SJM виявляють в одного із 80 000-160 000 хворих на ХНЕХ [17]. За підрахунками деяких вчених, це захворювання рідко діагностують – тільки в 2% випадків. Фермент лізосомальна кисла ліпаза (ЛКЛ) кодується геном LIPA [3, 17]. Внаслідок мутації в цьому гені активність ЛКЛ в клітині знижується, що призводить до накопичення ефірів ХС і меншою мірою тригліцеридів (ТГ) всередині клітин, переважно гепатоцитів, що продукують стероїди клітин надниркових залоз, слизової оболонки кишечника і системи фагоцитів. Ген LIPA розташований в хромосомі 10. ХНЕХ успадковується за аутосомно-рецесивним типом.
При ХНЕХ активність ЛКЛ становить 1-12%, тому накопичення ефірів ХС в організмі відбувається поступово, так само, як і формування клінічної картини, через що захворювання зазвичай діагностують у дітей більш старшого віку і у дорослих. Однак ретроспективно можна встановити, що у більшості хворих збільшення печінки і/або активності трансаміназ виявляють ще в дошкільному віці.
Перші симптоми ХНЕХ з’являються приблизно у віці 5 років, але їх маніфестація можлива і в значно більш пізньому віці. Однак встановити правильний діагноз більшості таких пацієнтів вдається лише до 20 років. Практично у всіх хворих на ХНЕХ спостерігається гепатомегалія, зумовлена відкладенням ефірів ХС у гепатоцитах і клітинах Купфера. У 74% хворих виявлено спленомегалію, яка також розвинулася внаслідок відкладення ефірів ХС у макрофагах селезінки [7].
Серед відхилень у показниках за даними біохімічного аналізу крові звертають на себе увагу гіперхолестеринемія, яку виявляють у 81% хворих на ХНЕХ, та збільшення концентрації ХС ЛПНЩ, що спостерігається майже у всіх хворих [2]. У багатьох пацієнтів знижений рівень ХС ЛПВЩ у крові. Збільшення активності аспартатамінотрансферази (АСТ) і аланінамінотрансферази (АЛТ) спостерігається практично у всіх хворих і часто є однією з перших ознак захворювання, причому активність цих ферментів у крові варіює в широкому діапазоні та іноді перевищує верхню межу норми у 100 разів [3].
Через залучення в патологічний процес кишечника хворі скаржаться на діарею, біль у животі, стеаторею. Внаслідок раннього розвитку атеросклерозу у таких пацієнтів у ранньому віці часто спостерігаються ішемічна хвороба серця, аневризма аорти та гостре порушення мозкового кровообігу.
Кальцифікація наднирників описана лише у 5% хворих на ХНЕХ [2].
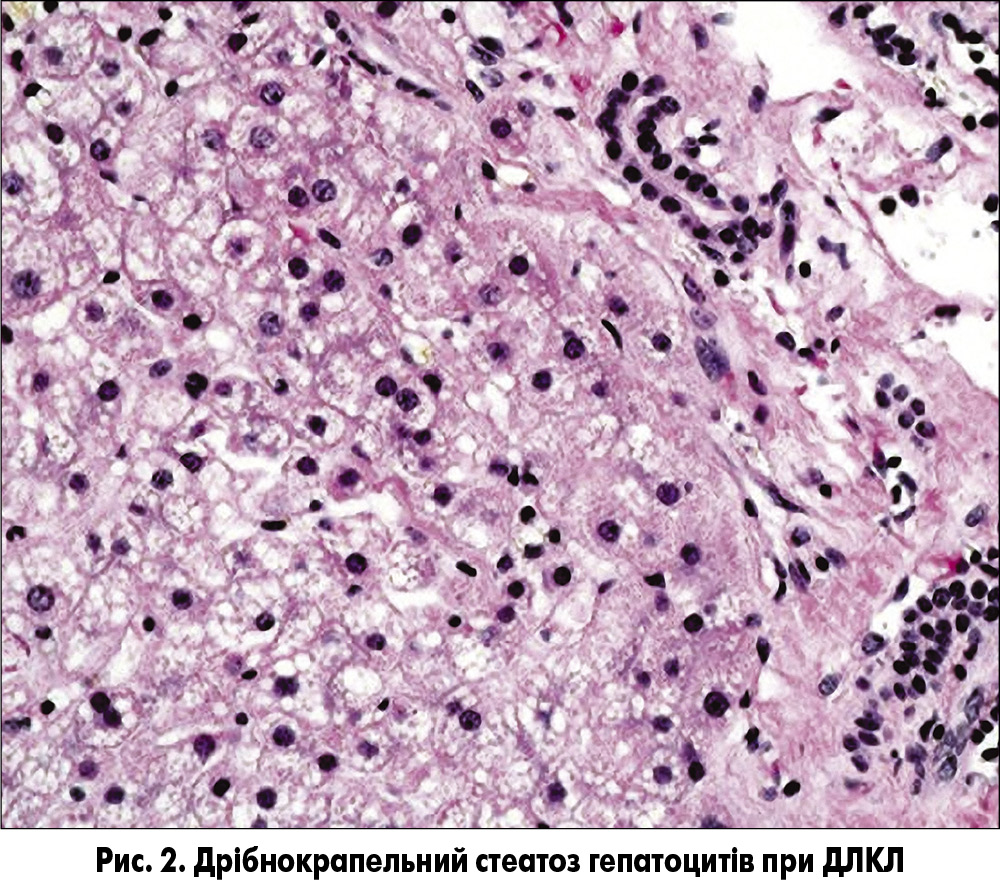
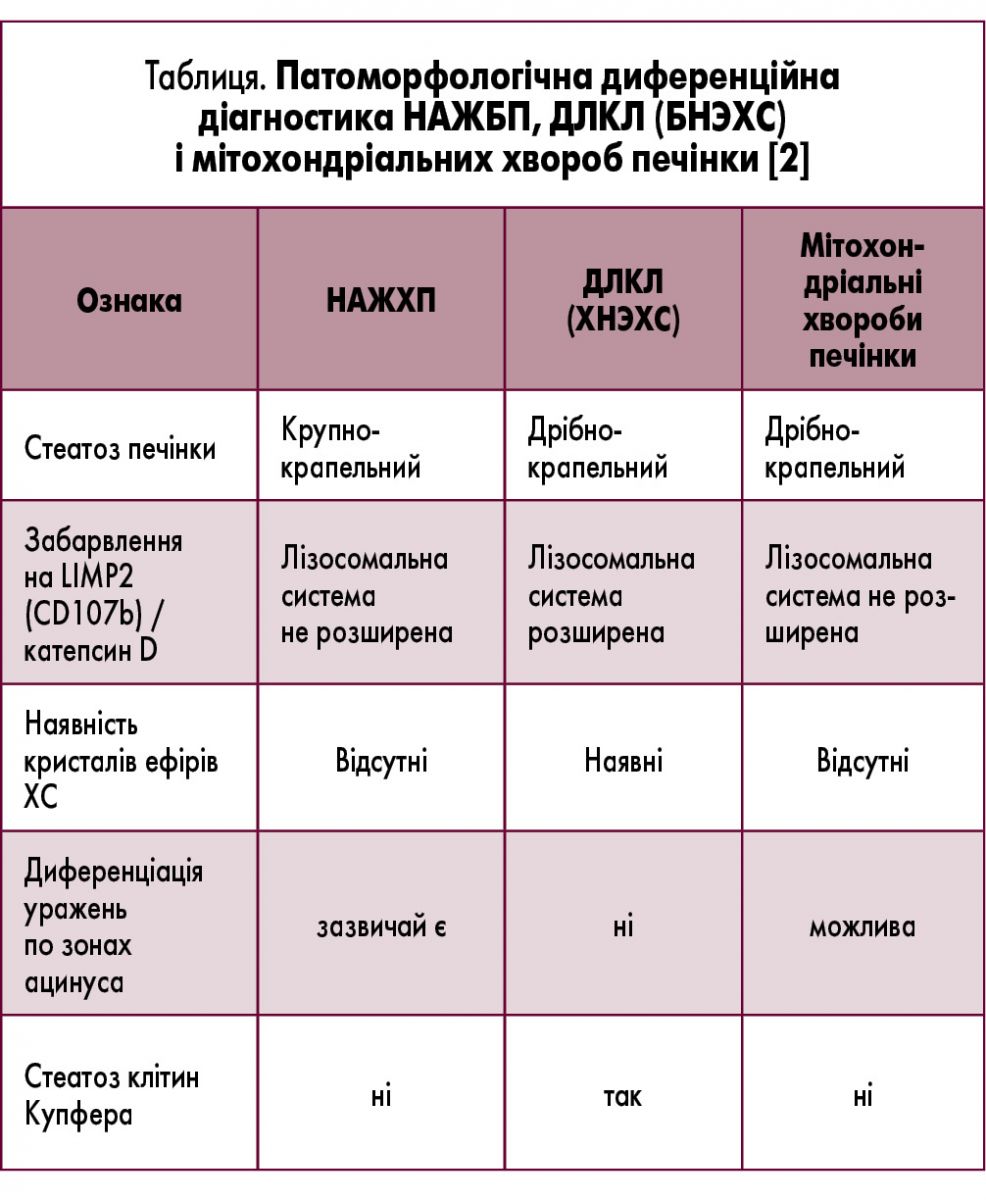
При гістологічному дослідженні тканини печінки у пацієнтів із ДЛКЛ визначають дрібнокрапельний стеатоз гепатоцитів (рис. 2), рівномірно розподілений по всьому печінковому ацинусу, і стеатоз клітин Купфера («пінисті» клітини). Дрібнокрапельний стеатоз гепатоцитів може спостерігатися і при деяких видах ураження печінки, спричиненого прийомом ліків, і при мітохондріальних хворобах (наприклад, дефекти білків дихального ланцюга, β-окислення жирних кислот), однак при цих захворюваннях відсутні «пінисті» макрофаги і кристали ЕХС. Важливе значення має диференційна діагностика з НАЖХП (табл.). Виявлення кристалів ефірів ХС у зрізах, фіксованих заморожуванням, за допомогою поляризаційного мікроскопа вважається гістологічним ознакою, яка є патогномонічною для ДЛКЛ [18].
Для точної патоморфологічної диференційної діагностики ДЛКЛ та інших хвороб, що супроводжуються дрібнокрапельним стеатозом печінки, запропоновані спеціальні імуногістохімічні забарвлення на білки лізосом, наприклад на LIMP2 (Lysosomal-associated membrane protein 2 – мембранний білок лізосом 2 або CD107b) і катепсини D.
 У 50% хворих із ДЛКЛ виявлено синусоїдальний, портальний або септальний фіброз печінки без розвитку цирозу, у 29% розвивався цироз, причому у 15% при серійній біопсії визначено перехід фіброзу в цироз [7].
У 50% хворих із ДЛКЛ виявлено синусоїдальний, портальний або септальний фіброз печінки без розвитку цирозу, у 29% розвивався цироз, причому у 15% при серійній біопсії визначено перехід фіброзу в цироз [7].
За наявності ознак стеатозу печінки необхідно проводити диференційну діагностику АЖХП і НАЖХП, а також ХНЕХ. Про наявність ХНЕХ свідчать початок захворювання у віці до 25 років, відсутність ЦД та ожиріння. Поєднання стеатозу печінки, дисліпідемії типу II b і спленомегалії у дітей і пацієнтів молодого віку з нормальною масою тіла – характерна ознака ХНЕХ. У тому випадку, якщо у хворого на НАЖХП корекція маси тіла і глікемії не призвели до регресії стеатозу печінки, потрібно виключити ХНЕХ. Це захворювання слід запідозрити у пацієнтів молодого віку з резистентною до статинів гіперхолестеринемією, гепатомегалією і зростанням рівнів АЛТ і АСТ у сироватці крові [2].
Загальновизнані діагностичні критерії ДЛКЛ (ХНЕХ) відсутні. На підставі наявних даних можна припустити, що діагноз ДЛКЛ (ХНЕХ) може бути встановлений за наявності принаймні однієї з перелічених ознак: зниження активності ЛКЛ, специфічних мутацій гена LIPA, характерної патоморфологічної картини (наявність кристалів ефірів ХС, стеатоз клітин Купфера, експансія лізосомної системи, що збігається з місцем відкладення ліпідів), характерної МР-спектрограми печінки [2].
Активність ЛКЛ можна визначати в біоптаті печінки, лейкоцитах крові та культурі фібробластів. У всіх випадках ДЛКЛ їх активність становила менше 16% від норми [1].
Призначена терапія препаратом себеліпаза-альфа належить до методів патогенетичного лікування при ДЛКЛ [5]. Статини та інші препарати, які знижують рівень ХС, істотно не впливають на вираженість жирової інфільтрації печінки [11, 19].
Різні типи мукополісахаридозів трапляються серед дорослих пацієнтів, які хворіють від народження і мають ознаки ураження печінки та селезінки [1]. Це хворі з вираженою клінічною картиною, що включає тяжкі ураження м’язово-кісткової системи, ЦНС, органів зору та слуху. Батьки таких дітей рано звертаються до педіатрів і генетиків, тому діагноз часто встановлюється у дитинстві. При деяких хворобах цієї групи тривалість життя може становити понад 70 років, наприклад при синдромі Моркіо (мукополісахаридозі IV типу) [1, 20].
У практиці терапевта і гастроентеролога можуть зустрічатися також дорослі пацієнти з глікогенозом [6].
Глікогеноз ІІІ типу (хвороба Корі)
Пов’язаний з мутаціями структурного гена цитозольної аміло-1, 6-глюкозидази, що експресуються у багатьох тканинах: печінці, м’язах, еритроцитах. Ген картований на хромосомі 1р21. ІІІ тип глікогенозу успадковується за аутосомно-рецесивним типом.
Хвороба Корі є дуже рідкісною для європейської популяції, але серед євреїв-сефардів (вихідців із Північної Африки) її частота становить 1:5400 новонароджених.
Фермент аміло-1,6-глюкозидаза, з одного боку, перетворює ліміт-декстрин на глікоген із зовнішніми ланцюгами нормальної довжини і, з іншого боку, вивільняє глюкозу. Недостатність ферменту призводить до порушення глікогенолізу і накопичення у тканинах молекул глікогену аномальної форми з укороченими зовнішніми ланцюгами. Порушення глікогенолізу супроводжується гіпоглікемією, лактат-ацидозом, гіперкетонемією [1].
При патоморфологічному дослідженні у печінці, м’язах і серці відзначаються накопичення глікогену та аномалія структури глікогену. Гістологічно виявляються великі набряклі фібрили, які зазнали вакуолізації. Гепатоцити вакуолізовані та виглядають пінистими, а в портальних трактах відзначаються фіброз і круглоклітинна інфільтрація.
Хвороба Корі починає розвиватися у пацієнтів у віці від 6 міс до 3 років. Її характерні симптоми – значна гепатомегалія, відставання в рості, гіпотрофія, «лялькове» обличчя, локальні відкладення жиру, шкірні ксантоми, типові – лактат-ацидоз із гіперкетонемією при голодуванні. Гепатомегалія з дисфункцією печінки має тенденцію до зникнення в постпубертатний період. Хворі зазвичай доживають до дорослого віку. У дорослих пацієнтів домінує міопатія з прогресуючою м’язовою слабкістю при фізичному навантаженні (проявом якої іноді є хитка хода), гіпотрофія м’язів кінцівок [1].
Діагностика цього захворювання проводиться на підставі клінічної картини та даних лабораторних досліджень: зниження активності аміло-1,6-глюкозидази і відкладення глікогену зміненої структури в гепатоцитах і м’язах. У плазмі крові відзначається збільшення концентрації лактату, сечової кислоти, ХС і ТГ.
При лікуванні слід запобігати гіпоглікемії голодування і компенсувати дефіцит амінокислот.
Глікогеноз VI типу (хвороба Герса)
Глікогеноз VI типу (гепатофосфорилазна недостатність) – це глікогеноз, спричинений недостатністю фосфорилази печінки [1].
Фосфорилаза печінки каталізує фосфорилювання (розщеплення) глікогену з утворенням глюкозо-1-фосфату. Порушення цього механізму призводить до надмірного відкладення глікогену в печінці.
Глікогеноз VI типу дебютує зазвичай на першому році життя. Його основні симптоми – значне збільшення печінки внаслідок інфільтрації гепатоцитів глікогеном, затримка росту, «лялькове» обличчя, гіперліпідемія, гіперглікемія після внутрішньовенного введення галактози, підвищений вміст глікогену в еритроцитах [1].
Для підтвердження діагнозу глікогенозу і встановлення його типу в стаціонарі проводять біопсію печінки, м’язів (іноді – шкіри) з подальшим гістохімічним дослідженням; при цьому визначають вміст глікогену в тканинах і активність ферментів, задіяних у його обміні [1].
Лікування спрямоване на усунення обмінних порушень, в тому числі ацидозу. В деяких випадках доцільно застосовувати глюкагон, анаболічні гормони і кортикостероїди. При гіпоглікемії їжу необхідно вживати часто, вона має містити багато легкозасвоюваних вуглеводів. При м’язовій формі глікогенозу поліпшення спостерігається завдяки дотриманню дієти з високим вмістом білка, призначенню фруктози (по 50-100 г на день перорально), полівітамінів та препаратів аденозинтрифосфату [1].
При підозрі на ЛХН необхідно проводити консультацію генетика та курацію таких хворих у подальшому з позицій мультидисциплінарного підходу [1].
Таким чином, ЛХН мають бути включені до переліку диференційного діагнозу в пацієнтів з ураженнями печінки та/або селезінки у дорослих. Хвороба Гоше І типу, хвороба Німана-Піка (тип В і С) та ХНЕХ є найбільш поширеними з цієї великої групи захворювань, які, якщо їх вчасно діагностувати, можна успішно лікувати за допомогою замісної терапії.
Література
- Краснопольская К.Д. Наследственные болезни обмена веществ. – М., 2005. – С. 2-20.
- Маевская М.В., Ивашкин В.Т., Жаркова М.С. и др. Редкие формы неалкогольной жировой болезни печени: наследственный дефицит лизосомной кислой липазы // РЖГГК. – 2016. – № 3. – С. 41-51.
- Aslanidis C., Ries S., Fehringer P. et al. Genetic and biochemical evidence that CESD and Wolman disease are distinguished by residual lysosomal acid lipase activity // Genomics. – 1996. – Vol. 33. – P. 85-93.
- Bernstein D.L., Hulkova H., Bialer M.G. et al. Cholesteryl ester storage disease: Review of the findings in 135 reported patients with an underdiagnosed disease // J Hepatol. – 2013. – Vol. 58. – P. 1230-1243.
- Burton B.K., Balwani M., Feillet F. et al. A Phase 3 Trial of Sebelipase Alfa in Lysosomal Acid Lipase Deficiency // N Engl J Med. – 2015. – Vol. 373 (11). – P. 1010-1020.
- Cassiman D., Packman S., Bembi B. et al. Cause of death in patients with chronic visceral and chronic neurovisceral acid sphingomyelinase deficiency (Niemann-Pick disease type B and B variant): Literature review and report of new cases // Mol. Genet. Metab. – 2016. – Vol. 118 (3). – P. 206-213.
- Hulkova H., Elleder M. Distinctive histopathological features that support a diagnosis of cholesterol ester storage disease in liver biopsy specimens // Histopathology. – 2012. – Vol. 60. – P. 1107-1113.
- Grabowski G.A. Phenotype, diagnosis, and treatment of Gaucher’s disease // Lancet. – 2008. – Vol. 372 (9645). – P. 1263-1271.
- Grabowski G.A., Charnas L., Du H. Lysosomal acid lipase deficiencies: the Wolman disease/cholesteryl ester storage disease spectrum. In: Scriver Valle D., Beaudet A.L., Vogelstein B., Kinzler K.W., Antonarakis S.E., Ballabio A., editors. Metabolic and molecular bases of inherited disease – OMMBID. New York: McGraw-Hill; 2012 // www.ommbid.com.
- Grabowski G.A., Kacena K., Cole J.A. et al. Dose-response relationships for enzyme replacement therapy with imiglucerase/alglucerase in patients with Gaucher disease type 1 // Genet. Med. – 2009. – Vol. 11(2). – P. 92-100.
- Leone L., Ippoliti P.F., Antonicelli R. Use of simvastatin plus cholestyramine in the treatment of lysosomal acid lipase deficiency // J Pediatr. – 1991. – Vol. 119. – P. 1008-1009.
- McGovern M.M., Lippa N., Bagiella E. et al. Morbidity and mortality in type B Niemann-Pick disease // Genet. Med. – 2013. – Vol. 15(8). – P. 618-623.
- Patterson M.C., Hendriksz C.J., Walterfang M. et al. NP-C Guidelines Working Group Recommendations for the diagnosis and management of Niemann–Pick disease type C: An update // Mol. Genet. Metab. – 2012 doi:10.1016/j.ymgme.2012.03.012
- Poll L.W., Terk M.R. Skeletal aspects of Gaucher disease // Brit J Radiol. 2002; 75(Suppl. 1): A1-A42.
- Poznansky M.J., Hutchison S.K., Davis P.J. Enzyme replacement therapy in fibroblasts from a patient with cholesteryl ester storage disease // FASEB J. – 1989 – Vol. 3. – P. 152-156.
- Sidransky E., Pastores G.M., Mori M. Dosing enzyme replacement therapy for Gaucher disease: older, but are we wiser? // Genet. Med. – 2009. – Vol. 11(2). – P. 90-91.
- Scott S.A., Liu B., Nazarenko I. et al. Frequency of the cholesteryl ester storage disease common LIPA E8SJM mutation (c.894G>A) in various racial and ethnic groups // Hepatol. – 2013. – Vol. 58. – P. 958-965.
- Stitziel N.O., Fouchier S.W., Sjouke B. et al. Exome sequencing and directed clinical phenotyping diagnose cholesterol ester storage disease presenting as autosomal recessive hypercholesterolemia // Arterioscler. Thromb. Vasc. Biol. – 2013. – Vol. 33. – P. 2909-2914.
- Xu M., Liu K., Swaroop M., Porter F.D. et al. d-Tocopherol reduces lipid accumulation in Niemann Pick type C1 and Wolman cholesterol storage disorders // J Biol Chem. – 2012. – Vol. 287. – P. 39349-60.
- Yoshida H., Kuriyama M. Genetic lipid storage disease with lysosomal acid lipase deficiency in rats // Lab. Anim. Sci. – 1990. – Vol. 40. – P. 486-489.
Тематичний номер «Гастроентерологія. Гепатологія. Колопроктологія» № 4 (46), листопад 2017 р.
СТАТТІ ЗА ТЕМОЮ Гастроентерологія
Метаболічноасоційована жирова хвороба печінки (МАЖХП) є однією з найактуальніших проблем сучасної гепатології та внутрішньої медицини в цілому. Стрімке зростання поширеності ожиріння та цукрового діабету (ЦД) 2 типу в популяції призвело до істотного збільшення кількості хворих на МАЖХП, яка охоплює спектр патологічних станів від неускладненого стеатозу до алкогольної хвороби печінки та цирозу, що розвиваються на тлі надлишкового нагромадження ліпідів у гепатоцитах. ...
Інфекція Helicobacter pylori (H. pylori) офіційно визнана інфекційним захворюванням і включена до Міжнародної класифікації хвороб (МКХ) 11-го перегляду, тому рекомендовано лікувати всіх інфікованих пацієнтів. Проте, зважаючи на широкий спектр клінічних проявів, пов’язаних із гастритом, викликаним H. pylori, лишаються специфічні проблеми, які потребують регулярного перегляду для оптимізації лікування. ...
Відтворення майбутнього здорової нації – один з найважливіших сенсів існування теперішнього покоління. День боротьби з ожирінням нагадує нам про поширеність цього проблемного явища і важливість попередження його наслідків. Ожиріння може мати вплив на різні аспекти здоров'я, включаючи репродуктивне....
Вивчення клініко-патогенетичних особливостей поєднаного перебігу остеоартрозу (ОА) у хворих із метаболічними розладами, які характеризують перебіг метаболічного синдрому (МС), зокрема цукровим діабетом (ЦД) 2 типу, ожирінням (ОЖ), артеріальною гіпертензією (АГ), є актуальним, оскільки це пов’язано з неухильним збільшенням розповсюдженості цього захворювання, недостатньою ефективністю лікування, особливо за коморбідності з іншими захворюваннями, які патогенетично пов’язані з порушеннями метаболічних процесів. ...