



Прогрессирующая мышечная дистрофия Дюшенна: методы функциональной диагностики, электронейромиографическое исследование и оценка поражения сердечной мышцы
Многие нервно-мышечные заболевания сопровождаются поражением миокарда. Среди них наиболее распространенными являются прогрессирующие мышечные дистрофии (ПМД), в частности ПМД Дюшенна. Атрофический процесс при ПМД поражает и скелетные, и сердечную мышцы. Это связано с тем, что в этиологии и патогенезе заболеваний сердечной и скелетной мышц много общего, ведь, несмотря на существующие различия, эти мышцы обладают сходством в молекулярной, анатомической структуре и физиологии [3]. При ПМД Дюшенна 84-96% больных страдают патологией со стороны сердечно-сосудистой системы [1, 2]. Дилатационная кардиомиопатия (ДКМП), желудочковые аритмии, фибрилляция предсердий, нарушения атриовентрикулярной и внутрижелудочковой проводимости, а также внезапная сердечная смерть – хорошо известные проявления мышечной дистрофии Дюшенна.
 ПМД Дюшенна – наследственное рецессивное Х-сцепленное заболевание, с распространенностью 1 случай на 3600-6000 мальчиков, рожденных живыми, возникающее в результате мутации в гене дистрофина и характеризующееся поражением проксимальных групп мышц, кардиологическими, ортопедическими и респираторными осложнениями (рис. 1). Другими словами, это наследственное заболевание, которое возникает в результате дефектов гена (делеция, дупликация или точечная мутация гена), кодирующего белок дистрофин. Дистрофин, в свою очередь, локализован в плазматической мембране скелетных мышечных волокон и кардиомиоцитов. ПМД Дюшенна является наиболее тяжелой формой с манифестацией в возрасте 2-5 лет и прогрессирующим злокачественным течением: формированием вялых парезов, параличей и мышечных контрактур, что приводит в будущем к обездвиженности.
ПМД Дюшенна – наследственное рецессивное Х-сцепленное заболевание, с распространенностью 1 случай на 3600-6000 мальчиков, рожденных живыми, возникающее в результате мутации в гене дистрофина и характеризующееся поражением проксимальных групп мышц, кардиологическими, ортопедическими и респираторными осложнениями (рис. 1). Другими словами, это наследственное заболевание, которое возникает в результате дефектов гена (делеция, дупликация или точечная мутация гена), кодирующего белок дистрофин. Дистрофин, в свою очередь, локализован в плазматической мембране скелетных мышечных волокон и кардиомиоцитов. ПМД Дюшенна является наиболее тяжелой формой с манифестацией в возрасте 2-5 лет и прогрессирующим злокачественным течением: формированием вялых парезов, параличей и мышечных контрактур, что приводит в будущем к обездвиженности.
Клинические проявления ПМД Дюшенна
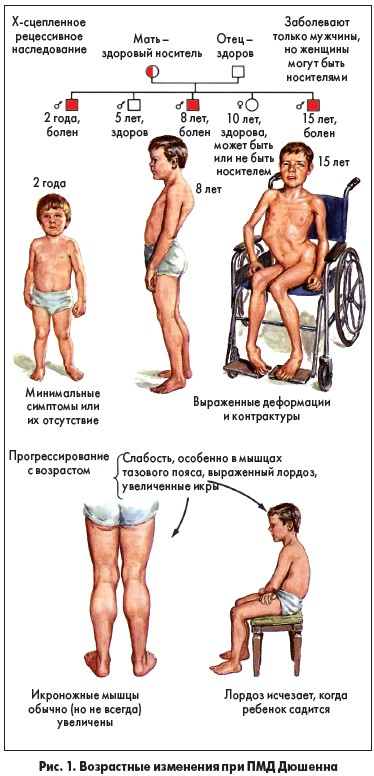 Ввиду того что ПМД Дюшенна связана с мутациями в гене, который находится на Х-хромосоме, этим заболеванием страдают только мальчики. Описания данной патологии у девочек крайне редки.
Ввиду того что ПМД Дюшенна связана с мутациями в гене, который находится на Х-хромосоме, этим заболеванием страдают только мальчики. Описания данной патологии у девочек крайне редки.
Основным симптомом является мышечная слабость, которая в первую очередь связана с атрофией скелетных мышц.
Как было указано выше, первые симптомы заболевания возникают на втором-третьем году жизни и обычно становятся очевидными к 5-6 годам. Сначала происходит поражение мышц конечностей, прежде всего ног, позже мышечная слабость возникает в других частях тела. Мышечная слабость становится настолько явной, что ребенок с трудом встает с пола, его походка изменяется и приобретает тип «утиной», ребенок прибегает к так называемым приемам Говерса – «взбирание по самому себе» и «подъем лесенкой». Еще один характерный признак этой болезни – «мнимая гипертрофия» мышц из-за появления в них жировой и соединительной тканей; кажется, что мышцы сильные и развитые, но на самом деле они очень слабые. Отмечается выпадение сухожильных рефлексов, коленные рефлексы исчезают на ранних стадиях заболевания, позднее исчезают рефлексы с двухглавой и трехглавой мышц плеча, ахилловы рефлексы обычно длительное время остаются сохранными. По мере прогрессирования заболевания пациенты теряют способность ходить, у них возникают деформации скелета, нарушается подвижность из-за контрактур [18-20].
Облигатным признаком развернутой стадии ПМД Дюшенна является гипертрофическая, или дилатационная, кардиомиопатия, которая сопровождается нарушениями ритма сердца, расширением его границ, явлениями сердечной недостаточности (СН). Кардиомиопатия – наиболее частая причина летального исхода при ПМД Дюшенна [4, 5]. К летальности приводит также дыхательная недостаточность, которая провоцируется интеркуррентными инфекциями или аспирацией. Больные погибают на втором-третьем десятилетии жизни.
Диагностика
При биохимическом анализе крови выявляют повышение активности печеночных ферментов (АлАТ, АсАТ) и значительное – в сотни и тысячи раз – повышение активности креатинфосфокиназы. Это отражает процесс разрушения мышечной ткани.
Основным методом для подтверждения диагноза является ДНК-диагностика, довольно сложная и дорогая. В некоторых странах обсуждается возможность проведения массового скрининга на данное заболевание.
Поэтому при постановке диагноза мы стараемся использовать дополнительные методы. Один из них – электромиография (ЭМГ). Этот метод позволяет подтвердить тот факт, что в основе заболевания лежит первично-мышечный тип изменений, а периферическая нервная система при этом совершенно интактна [6]. ЭМГ-исследование на сегодняшний день является одним из основных методов в оценке структурно-функционального состояния мышц и диагностике нервно-мышечных заболеваний. ЭМГ используют для дифференциальной диагностики между первичными (мышечными) и вторичными (невральными) мышечными дистрофиями.
ЭМГ (стимуляционная) – это комплекс методов оценки функционального состояния нервно-мышечной системы, основанный на регистрации и анализе биоэлектрической активности мышц и периферических нервов. К основным заболеваниям, при которых ЭМГ-диагностика является наиболее информативной и часто используемой, относятся первично-мышечная патология (ПМД, полимиозит, миозит), невральная патология (радикулопатии, плексопатии, невропатии (травматические; компрессионно-ишемические; полиневропатии – воспалительные, наследственные, дисметаболические, токсические), заболевания периферического двигательного мотонейрона (полиомиелит, боковой амиотрофический склероз).
Электронейромиография (ЭНМГ) как диагностический метод включает в себя две основные методики – стимуляционную и игольчатую. В основе стимуляционной ЭНМГ (рис. 2) лежит регистрация сумарного ответа мышцы или нерва в ответ на стимуляцию импульсом электрического тока. Методы стимуляционной ЭНМГ позволяют исследовать проводящую функцию моторных и сенсорных аксонов периферических нервов, а также функциональное состояние нервно-мышечной передачи. Также сюда относят исследование поздних ответов, таких как F-волна (данный метод позволяет выявить поражение нейромоторного аппарата на ранней субклинической стадии заболевания) и Н-рефлекс (является аналогом ахиллового рефлекса, посредством которого можно оценить порог возбудимости мотонейронов поясничного утолщения). С помощью игольчатой ЭМГ (рис. 3) определяют функциональное состояние мышцы, наличие или отсутствие текущего процесса в ней, степень и выраженность денервации, выраженность и эффективность реиннервации. В основе этого метода лежит определение параметров потенциалов двигательных единиц (ПДЕ), которое включает анализ длительности, фазности и амплитуды, а также анализ спонтанной активности ДЕ и мышечных волокон.
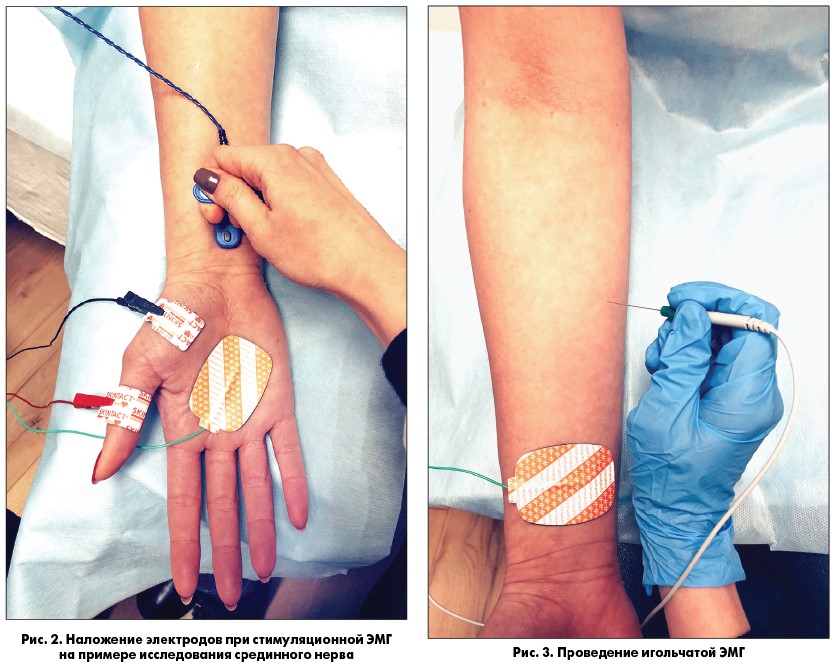
В диагностике ПМД Дюшенна стимуляционная ЭНМГ особой роли не играет. При исследовании проводящей функции моторных нервов выявляется снижение амплитуды М-ответа, соответствующее снижению силы мышцы и выраженности атрофии; остальные параметры остаются в норме до поздней стадии заболевания. Более информативной является игольчатая ЭМГ. Она и служит основным методом исследования при подозрении на ПМД. Выполнение данной методики позволяет выявить первично-мышечный тип изменений потенциалов ДЕ (уменьшение длительности и снижение амплитуды ПДЕ) и спонтанную активность мышечных волокон (в виде потенциалов острых волн – ПОВ, потенциалов фибрилляции – ПФ), указывающих на степень активности процесса в каждой конкретной мышце. Спонтанная активность, регистрируемая при ПМД, всегда значительно выражена, что отличает ПМД от других наследственных первично-мышечных заболеваний. Она наблюдается в самых начальных стадиях заболевания, когда наряду с ПФ выявляются ПОВ и разряды высокой частоты [6].
Поражение сердца при ПМД Дюшенна
Несмотря на то что проблема вовлечения сердца в патологический процесс при миодистрофии Дюшенна активно изучается с 1980-х гг. [21, 22], до сих пор уровень выявляемости сердечно-сосудистых нарушений и профилактика тяжелых осложнений на ранних стадиях остаются очень низкими. Причиной летального исхода при большинстве форм ПМД является вовлечение в патологический процесс сердечной мышцы [23, 24].
Все это обусловливает необходимость дальнейшей разработки вопросов ранней диагностики кардиологических нарушений при различных формах ПМД с целью определения тактики лечения, профилактики развития тяжелых сердечно-сосудистых нарушений, улучшения качества и продолжительности жизни больных.
Нарушения со стороны сердца проявляются в виде ишемических и метаболических изменений миокарда, перегрузки левого желудочка (ЛЖ), нарушений проводимости и сердечного ритма (в виде внутрисердечных блокад и аритмий), а также развития внезапной сердечной смерти. Структурные изменения сердца с дилатацией ЛЖ или гипертрофией встречаются при миотонической дистрофии приблизительно у 20% больных, систолическая дисфункция ЛЖ отмечается в 14% случаев [7, 8], но клинически очевидная СН встречается редко – всего у 2% больных. Кардиомиопатия у детей с миодистрофией Дюшенна развивается на фоне гипо- и адинамии, поэтому не проявляется СН I и IIA стадии и не провоцирует жизнеугрожающих нарушений ритма и проводимости сердца, что приводит к поздней диагностике.
На основании анализа данных многих исследований можно с уверенностью сказать, что пациенты с миодистрофией Дюшенна должны в обязательном порядке проходить кардиологическое обследование и обследование дыхательной системы на ранних этапах заболевания.
Скрининг для выявления сердечной патологии необходимо проводить всем детям с нервно-мышечными заболеваниями, при которых существует риск вовлечения в патологический процесс сердца. Рекомендации относительно времени и частоты проведения первичного и последующих обследований были разработаны Американской академией педиатров совместно с экспертами в данной предметной области и основываются на времени начала и клиническом течении поражения сердца при отдельных нервно-мышечных заболеваниях. Скрининг должен включать тщательный сбор анамнеза, физикальное обследование на предмет признаков и симптомов СН или аритмии [9]. Иногда установить наличие клинически значимой СН у пациентов с генерализованной мышечной слабостью бывает трудно вследствие присутствия симптомов, которые могут имитировать признаки СН. Необходимо фиксировать такие общие жалобы, как одышка, слабость, утомляемость, нарушение сна, снижение аппетита, потеря массы тела, поскольку они в том числе могут являться и симптомами недостаточности кровообращения. Головокружение или синкопальные состояния, ощущение перебоев в работе сердца, чувство учащенного сердцебиения могут быть признаками нарушения ритма сердца.
При постановке диагноза в скрининг при выявлении патологии со стороны сердца необходимо включать проведение инструментальных и лабораторных исследований.
Электрокардиография (ЭКГ) – важный инструмент скрининга для выявления нарушений ритма и проводимости сердца, гипертрофии или дилатации желудочков [10]. Проведение эхокардиографии считается золотым стандартом диагностики структурно-функциональных нарушений миокарда и способствует выявлению патологии сердца на доклиническом этапе [11].
Очень часто – в зависимости от уровня поражения сердца – требуется выполнение дополнительного обследования. При заболеваниях, сопровождающихся выраженными нарушениями ритма, рекомендуется проведение суточного мониторирования ЭКГ. Кроме того, для оценки степени фиброза миокарда, воспалительных изменений, патологии перикарда у пациентов могут применяться такие неинвазивные методы диагностики, как магнитно-резонансная или компьютерная томография сердца с контрастированием [12, 13].
Лечение больных с нарушениями ритма и проводимости сердца включает антиаритмическую терапию, имплантацию искусственного водителя ритма (электрокардиостимулятора), кардиовертера-дефибриллятора. Больные с ремоделированием миокарда, нарушениями систолической и/или диастолической функции миокарда и хронической СН (ХСН) должны получать лечение в соответствии с рекомендациям по терапии ХСН [14]. В настоящее время рассматривается вопрос о начале превентивной терапии патологии сердечно-сосудистой системы больных с нервно-мышечными болезнями в детском возрасте. Согласно данным Педиатрического реестра кардиомиопатии шестилетняя выживаемость у пациентов с мышечной дистрофией Дюшенна после установления диагноза ДКМП составляет 58%, а уровень выживаемости без трансплантации сердца – 55% [15]. Данные 10-летнего наблюдения указывают на более низкий уровень смертности среди пациентов, которым согласно рандомизированному отбору были изначально назначены ингибиторы ангиотензинпревращающего фермента (ИАПФ) [16]. Увеличение выживаемости по сравнению с данными литературы отмечено в исследованиях у пациентов с ПМД Дюшенна, получавших комбинированную терапию ИАПФ и β-блокаторами. Также у части пациентов выявлена нормализация функции и размеров ЛЖ [17].
Лечение
На сегодняшний день в Украине не существует специфического лечения мышечной дистрофии. Все лечение симптоматическое и направлено на поддержание жизненно важных систем и функций организма. В то же время за рубежом уже зарегистрирован препарат, стимулирующий выработку дистрофина. Также запатентована методика пересадки костного мозга от отца к сыну, что, к сожалению, не восстанавливает утраченные способности мышц, однако предотвращает дальнейшее их разрушение.
Хочется верить, что в ближайшем будущем дети в Украине смогут получать специфическое лечение, которое позволит улучшить качество их жизни и продлить ее.
Литература
- Ishikawa Y. Cardiovascular considerations in the menegement of neuromuscular disease / Y. Ishikawa, J.R. Bach // Seminars in neurology, 1995; 15: 93-108.
- Nigro G. Dilated cardiomyopathy of muscular dystrophy. A multifaceted approach to management / G. Nigro, G. Lucia, M.D. Comi, L. Politano // Seminars in neurology, 1995; 15: 90-92.
- Finsterer J., Stullberger C., Wahbi K. Cardiomiapathy in neurological disorders. Cardiovasc. Pathol. 2013. P. 389-400.
- Грознова О.С., Тренева М.С. Генетические аспекты возникновения жизнеугрожаемых состояний у больных миопатией // Российский вестник перинатологии и педиатрии, 2011. С. 38-41.
- Politano L., Nigro G. Treatment of distrophinopathic cardiomyopathy: review of the literature and personal results. Acta Myologica. 2012. P. 24-30.
- Касаткина Л.Ф., Гильванова О.В. Наследственные первично-мышечные заболевания. Электромиографические методы исследования в диагностике нервно-мышечных заболеваний, игольчатая электромиография. 2010. С. 285-296.
- Babuty D., Fauchier L., Tena-Carbi D., et al. It is possible to identify infrahissian cardiac conduction abnormalities in myotonic dystrophy by non-invasive methods. Heart, 1999; 82: 634-7.
- Bhakta D., Lowe M.R., Groh W.J. Prevalence of structural cardiac abnormalities in patients with myotonic dystrophy type I. Am. Heart J. 2004; 147: 224-7.
- Melacini P., Buja G., Fasoli G., et al. The natural history of cardiac involvement in myotonic dystrophy: an eight-year follow-up in 17 patients. Clin. Cardiol. 1988; 11: 231-8.
- Stollberger C., Winkler-Dworak M., Blazek G., Finsterer J. Association of electrocardiographic abnormalities with cardiac findings and neuromuscular disorders in left ventricular hypertrabeculation / non-compaction. Cardiology, 2007; 107: 374-9.
- Mertens L., Ganame J., Claus P., et al. Early regional myocardial dysfunction in young patients with Duchenne muscular dystrophy. J. Am. Soc. Echocardiogr. 2008; 21: 1049-54.
- Silva M.C., Meira Z.M., Gurgel Giannetti J., et al. Myocardial delayed enhancement by magnetic resonance imaging in patients with muscular dystrophy. J. Am. Coll. Cardiol. 2007; 49: 1874-9.
- Kazanegra R., Cheng V., Garcia A., et al. A rapid test for B-type natriuretic peptide correlates with falling wedge pressures in patients treated for decompensated heart failure: a pilot study. J. Card. Fail. 2001; 7 (1): 21-9.
- Басаргина Е.Н., Архипова Е.Н., Жарова О.П. Типичные ошибки при лечении хронической сердечной недостаточности со сниженной систолической функцией у детеи // Фарматека, 2014; 1: 55-62.
- Connuck D.M., Sleeper L.A., Colan S.D., et al. Characteristics and outcomes of cardiomyopathy in children with Duchenne or Becker muscular dystrophy: a comparative study from the Pediatric Cardiomyopathy Registry. Am. Heart J. 2008; 155: 998-1005.
- Duboc D., Meune C., Pierre B., et al. Perindopril preventive treatment on mortality in Duchenne muscular dystrophy: 10 years’ followup. Am. Heart J. 2007; 154: 596-602.
- Rhodes J., Margossian R., Darras B.T., et al. Safety and efficacy of carvedilol therapy for patients with dilated cardiomyopathy secondary to muscular dystrophy. Pediatr. Cardiol. 2008; 29: 343-51.
- Бадалян Л.О. Наследственные заболевания нервно-мышечной системы. – М.: Медицина, 1974. – С. 4-25.
- Божинова С., Гилабова Г. Миопатия. – София: Медицина и физкультура, 1977. – С. 7-77.
- Дубенко Е.Г. Нервные болезни. – К.: Здоровье, 2001. – С. 413-425.
- Осетров А.С., Малкова А.А., Осипова Е.В. и др. Нервно-мышечные заболевания: клиника, диагностика, лечение. – Ижевск, 2014. – С. 76.
- Грознова О.С., Руденская Г.Е., Адян Т.А. Поражение сердца при наследственных нервно-мышечных заболеваниях у детей // Российский вестник перинатологии и педиатрии, 2014; 59 (2): 35-42.
- Cotta A. Common recessive limb girdle muscular dystrophies differential diagnosis: why and how? Arq. Neuropsiquiatr. 2014; 72 (9): 721-734.
- Ergul Y., Ekici B., Nisli K. Evaluation of the North Star Ambulatory Assessment scale and cardiac abnormalities in ambulant boys with Duchenne muscular dystrophy. J. Paediatr. Child. Health. 2012; 10: 1440-1754.
Тематичний номер «Кардіологія, Ревматологія, Кардіохірургія» № 2 (56) Квітень 2018 р.
СТАТТІ ЗА ТЕМОЮ Кардіологія
Дисліпідемія та атеросклеротичні серцево-судинні захворювання (АСССЗ) є провідною причиною передчасної смерті в усьому світі (Bianconi V. et al., 2021). Гіперхолестеринемія – третій за поширеністю (після артеріальної гіпертензії та дієтологічних порушень) фактор кардіоваскулярного ризику в світі (Roth G.A. et al., 2020), а в низці європейських країн і, зокрема, в Польщі вона посідає перше місце. Актуальні дані свідчать, що 70% дорослого населення Польщі страждають на гіперхолестеринемію (Banach M. et al., 2023). Загалом дані Польщі як сусідньої східноєвропейської країни можна екстраполювати і на Україну....
Інколи саме з цього перерізу вдається візуалізувати тромбоемболи в основних гілках легеневої артерії або вегетації на стулках легеневого клапана (що трапляється надзвичайно рідко). Нахиливши датчик до самої верхівки серця, можна отримати її переріз по короткій осі, на якому, знову ж таки, порожнина лівого шлуночка має круглясту форму, а правого шлуночка – близьку до трикутника із виразною трабекулярністю (рис. 22.9). Розглядаючи зображення, також звертають увагу на те, що в нормі всі сегменти ЛШ скорочуються синхронно, не випереджаючи інші і не відстаючи. ...
Застосування статинів середньої інтенсивності в комбінації з езетимібом порівняно зі статинами високої інтенсивності окремо може забезпечити більше зниження рівня холестерину ліпопротеїнів низької щільності (ХС ЛПНЩ) у пацієнтів із нещодавнім ішемічним інсультом. Пропонуємо до вашої уваги огляд статті Keun-Sik Hong et al. «Moderate-Intensity Rosuvastatin Plus Ezetimibe Versus High-Intensity Rosuvastatin for Target Low-Density Lipoprotein Cholesterol Goal Achievement in Patients With Recent Ischemic Stroke: A Randomized Controlled Trial», опублікованої у виданні Journal of Stroke (2023; 25(2): 242‑250). ...
Артеріальна гіпертензія (АГ) сьогодні є одним із найпоширеніших серцево-судинних захворювань (ССЗ), що асоціюється з високим кардіоваскулярним ризиком, особливо в коморбідних пацієнтів. Навіть помірне підвищення артеріального тиску (АТ) пов’язане зі зменшенням очікуваної тривалості життя. До 40% хворих на АГ не підозрюють у себе недугу, бо це захворювання на початку може мати безсимптомний перебіг. Оптимальний контроль АТ є вагомим чинником профілактики фатальних серцево-судинних подій (ССП) для забезпечення якісного та повноцінного життя таких хворих. ...