



Роль ренін-ангіотензин-альдостеронової системи в регуляції артеріального тиску: огляд літератури
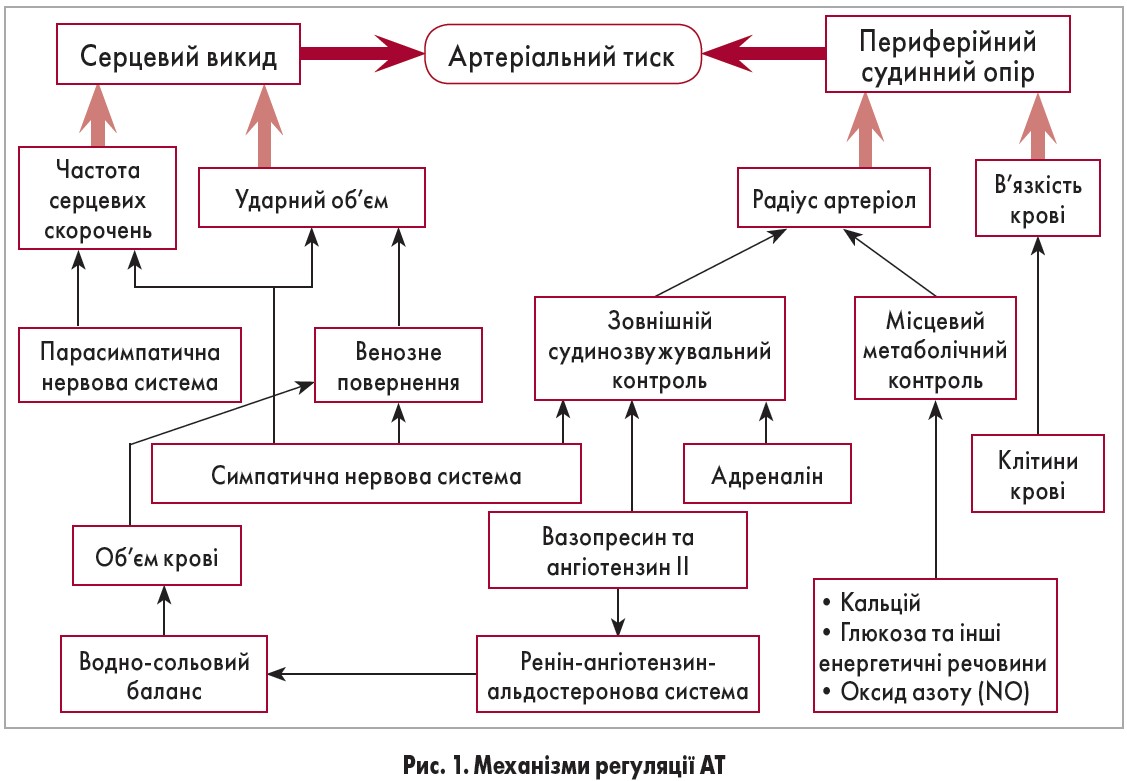
 Рівень артеріального тиску (АТ) насамперед визначається серцевим викидом і периферійним судинним опором. Ці чинники зазнають багатьох впливів, зокрема вегетативної нервової системи, об’єму і в’язкості циркулюючої крові, гормонів та біологічно активних сполук (рис. 1). При збільшенні серцевого викиду і/чи підвищенні периферійного судинного опору виникає артеріальна гіпертензія (АГ).
Рівень артеріального тиску (АТ) насамперед визначається серцевим викидом і периферійним судинним опором. Ці чинники зазнають багатьох впливів, зокрема вегетативної нервової системи, об’єму і в’язкості циркулюючої крові, гормонів та біологічно активних сполук (рис. 1). При збільшенні серцевого викиду і/чи підвищенні периферійного судинного опору виникає артеріальна гіпертензія (АГ).
Першовідкривачами ренін-ангіотензин-альдостеронової системи (РААС) можна вважати фінського фізіолога професора Роберта Адольфа Армана Тігерстедта (Tigerstedt R.) та його учня Пера Бергмана (Bergman P.), котрі відкрили ренін у лабораторії Каролінського університету в Стокгольмі. У 1898 році вчені продемонстрували, що внутрішньовенне введення екстракту з кортикального шару нирки кроликам-реципієнтам, яким було проведено нефректомію, підвищує АТ [1]. Проте ці результати не могли відтворити інші дослідники, тому відкриття реніну було проігнорованим та забутим на багато років.
Перший успішний експеримент проведено у США 1934 року під керівництвом Гаррі Голдблатта (Goldblatt H.). Бувши патологом, він помітив, що в осіб, які померли від АГ, спостерігається характерне звуження ниркових артерій. Припустивши, що ішемія нирки може викликати АГ, Г. Голдблатт винайшов срібну кліпсу, накладаючи яку на ниркові артерії собак довів, що часткове звуження обох ниркових артерій призводить до стійкого підвищення АТ за відсутності ниркової недостатності. При затисканні інших великих артерій – селезінкової чи стегнової – такого ефекту не було. У такий спосіб створено першу тваринну модель АГ (модель Голдблатта) та виявлено «внутрішню секрецію» нирки (ренін) [2].
У кінці 30-х років ХХ ст. історію відкриття РААС продовжили дві незалежні групи вчених – аргентинські науковці Едуардо Браун-Менендез (Braun-Menendez Е.), Хуан Фасціоло (Fasciolo J.), Луїс Федеріко Лелуар (Leloir L. F.) і Хуан Муньос (Munoz J.) з університету Буенос-Айреса, а також американські вчені Ірвін Пейдж (Page I. H.) та Оскар Хелмер (Helmer O. M.), які працювали у науковій лабораторії Елі Ліллі (Eli Lilly Research Laboratories) в Індіанаполісі, штат Індіана, США. Намагаючись очистити ренін, вони виявили несподіваний результат: чим вищою була чистота екстракту, тим меншою ставала його пресорна активність. Цей факт наштовхнув науковців на думку, що сам ренін не викликає підвищення АТ, а ймовірніше є протеолітичним ферментом, який перетворює пептид, що міститься у плазмі, на вазопресорну речовину. Тому очищення реніну від субстрату нівелювало його гіпертензивний ефект. У 1940 році було опубліковано дві праці, які вперше описували ангіотензин: американці назвали його «ангіотоніном» [3], а аргентинці – «гіпертензином» [4]. У 1958 вчені опублікували спільну працю, де і запропонували ввести до номенклатури об’єднану назву «ангіотензин» та його попередник «ангіотензиноген» [5].
Завдяки дослідженням Леонарда Скеггса (Skeggs L.) і співавт. ідентифіковано дві форми ангіотензину – І та ІІ, запропоновано назвати субстрат реніну ангіотензиногеном. Також з’ясовано, що утворення ангіотензину ІІ відбувається за участю ангіотензинперетворювального ферменту (АПФ) та описано роль РААС у патогенезі АГ [6-9].
Гіпотеза про взаємозв’язок між ангіотензином ІІ та альдостероном уперше запропонована професором Францом Гроссом (Gross F.) і підтверджена Джеймсом Девісом (Davis J. O.) [10, 11]. Доктор Джеймс Девіс провів багато досліджень, у яких вивчав виділення альдостерону клітинами клубочкової зони надниркових залоз під впливом ангіотензину ІІ та значення гіпоталамо-гіпофізарної осі в ендокринній регуляції [12, 13].
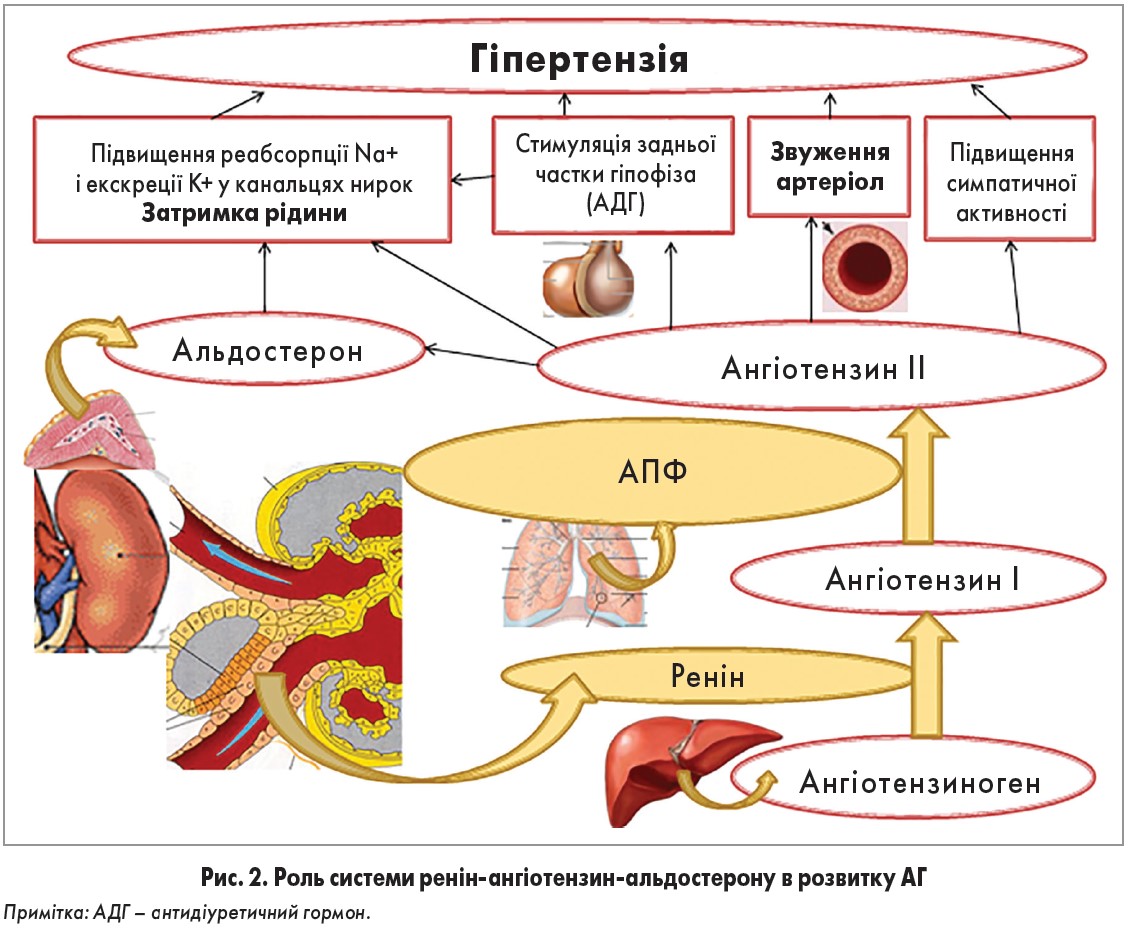
Отже, після багатьох років досліджень встановлено основні компоненти РААС та роль цієї системи в регуляції АТ і водно-сольового обміну (рис. 2).
Дослідження останніх років значно розширили уявлення про роль РААС в організмі (рис. 3). З’ясовано її функції у процесах диференціації та регенерації тканин, розвитку гіпертрофії, склерозу та запалення [14, 15, 16].
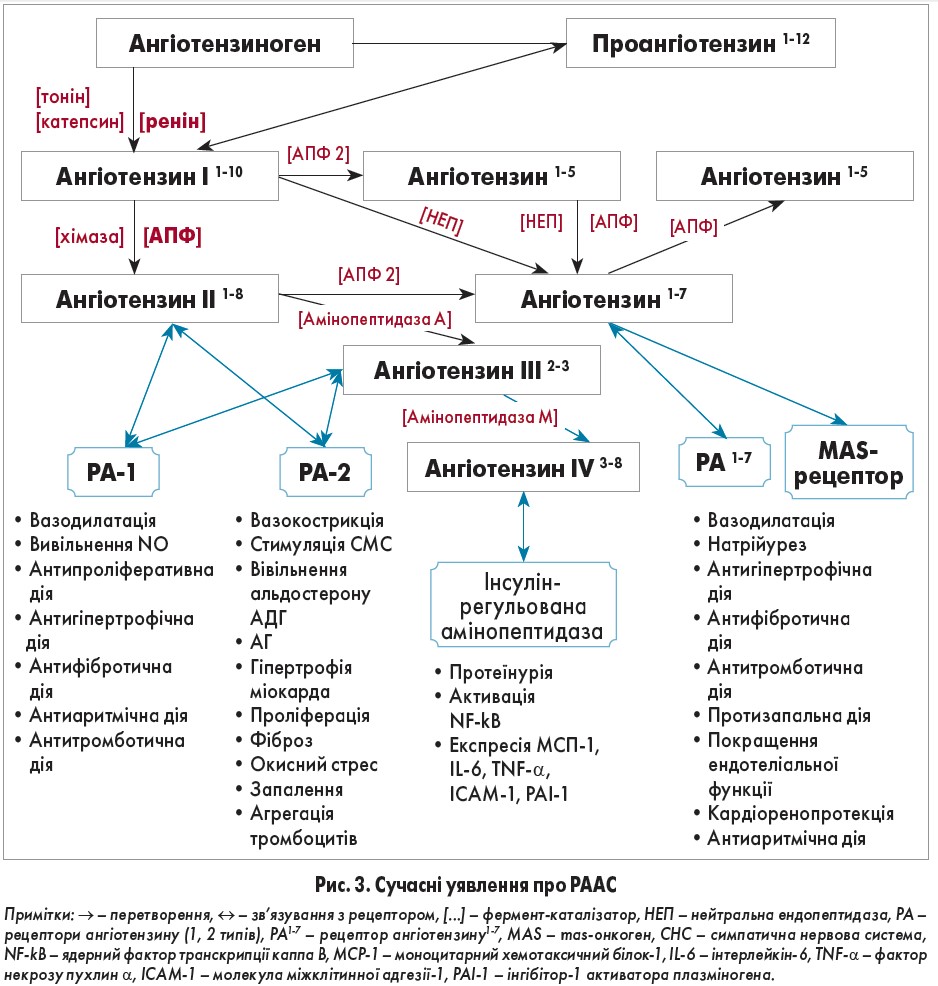
Розрізняють системну і тканинну РААС. Циркулюючі компоненти системної РААС виявляються в крові, мають ендокринний ефект та вiдповiдають за швидкі короткочаснi впливи, забезпечуючи регуляцію АТ і гемодинаміки при гiпертензивних кризах, гострій серцевій недостатності тощо. У системній РААС утворення ангіотензину ІІ та його кількість у крові визначається активністю реніну. Її діючими компонентами є циркулюючий проренін/ренін, що утворюється в нирках, АПФ, який присутній на зовнішніх поверхнях ендотеліальних клітин легень, та ангіотензиноген – сироватковий α2-глобулін, що синтезується у печінці. Реакції протеолізу відбуваються каскадно: ренін каталізує розщеплення ангіотензиногену з утворенням декапептиду ангіотензину І. Під час проходження через легеневий судинний шар ендотеліальний АПФ відщеплює від С-кінця ангіотензину І дипептид гістидин-лейцин. Як наслідок, утворюється системний сигнальний октапептид ангіотензину ІІ. Пролін – передостанній амінокислотний залишок у С-кінці його молекули блокує активність АПФ, забезпечуючи закінчення реакції. Тому ангіотензин ІІ є основним продуктом метаболізму ангіотензиногену у легеневому кровообігу [17, 18].
Дія тканинної РААС аутокринно/паракринна, відповідає за довготривалі ефекти на рiвнi органів. Вона ідентифікована у головному мозку, серці, судинах, нирках, надниркових залозах, підшлунковій залозі, кістковому мозку, жировій тканині, лімфатичній та репродуктивній системах [17, 19, 20]. Активація компонентів РААС пов’язана із структурно-функціональними змінами судин і серця, зокрема гіпертрофією та фіброзом, та є основним патогенетичним механізмом ураження органів-мішеней при АГ [21, 22]. Її складовими є ангіотензиноген, АПФ, ангіотензин та специфічні тканинні рецептори. Для утворення ангіотензину ІІ використовується локальний ангіотензин І. Цей процес каталізує АПФ, близько 90% якого зосереджено у тканинах. Альтернативними каталізаторами можуть бути серинові протеази (хімази, ендопептидази, катепсин G та калікреїн-подібні ферменти), що діють у серці, нирках і мозку [21, 19, 23, 16]. Для прикладу, у серці ідентифіковано фермент α-хімазу, що синтезується мастоцитами. Однією з її функцій є розщеплення ангіотензину І з утворенням ангіотензину ІІ [24]. Тканинний АПФ допомагає підтримувати рівновагу між вазодилатацією та вазоконстрикцією, стимуляторами та інгібіторами росту, про- та протизапальними чинниками, тромботичними та фібринолітичними шляхами у судинній стінці [25].
Нещодавно відкрито внутрішньоклітинну РААС, що діє за аутокринним типом та утворює ангіотензин ІІ для контролю росту клітин. Її функція у мітохондріях імовірно пов’язана з процесами тканинного дихання та виробництва NO [26].
Першим діючим компонентом РААС є проренін – препрогормон, який синтезується юкстагломерулярним апаратом нирок (міоепітеліальні клітини, що оточують аферентну артеріолу) та виводиться з клітин за допомогою сигнального пептиду [27]. Проренін також синтезується у яєчниках за умови їхньої гіперстимуляції; у плаценті під час вагітності; у макрогліальних клітинах Мюллера сітківки ока та збірних протоках нирок у хворих на цукровий діабет; а також у надниркових залозах і яєчках [28-31].
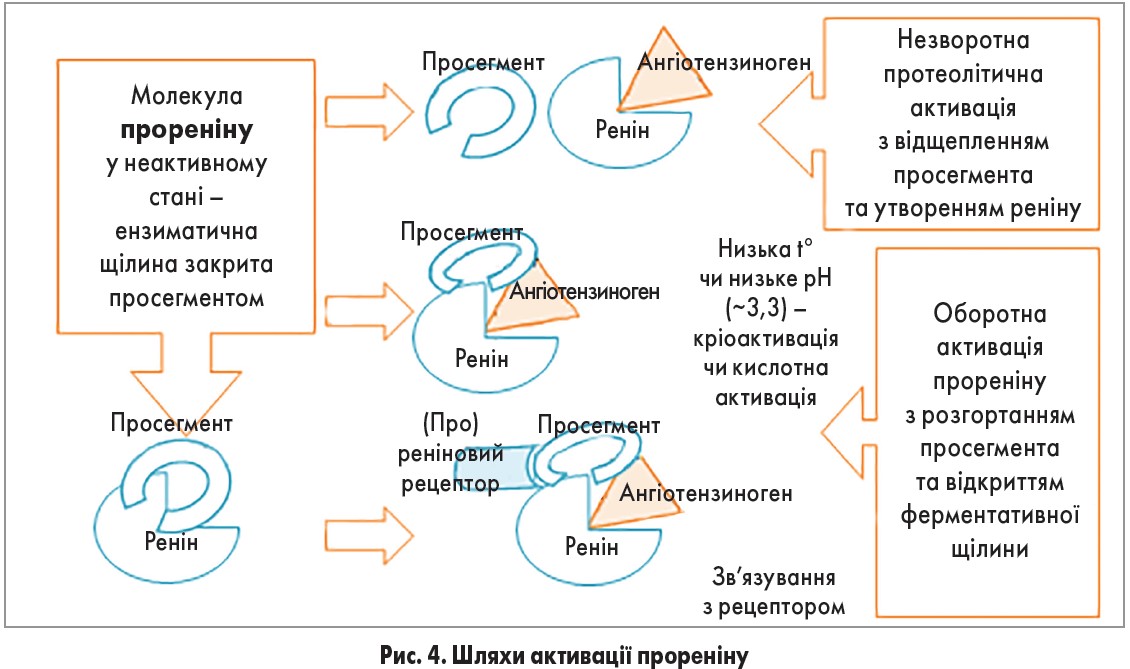
Молекула прореніну складається із двох гомологічних часток, розділених глибокою щілиною, яка містить активний центр. Ферментативна неактивність зумовлена тим, що щілину закриває просегмент – N-кінцевий пропептид, який складається із 43 амінокислотних залишків та перешкоджає зв’язуванню субстрату (ангіотензиногену) з активним сайтом [32]. Донедавна вважалося, що проренін є лише неактивним попередником реніну. Проте коли виявилось, що його концентрація у циркулюючій крові удесятеро перевищує концентрацію реніну (а при вагітності та діабеті у 100 разів), виникла думка, що проренін може відігравати в організмі власну біологічну роль. Згодом встановлено, що за певних умов (низькі значення температури чи рН) або після зв’язування з (про)реніновим рецептором ця сполука активується. За такої умови просегмент розгортається, відкриваючи ферментативну щілину, відбувається конформація молекули, унаслідок чого проренін стає ферментативно активним [33, 34]. Протеолітична активація прореніну відбувається у лізосомоподібних структурах клітин юкстагломерулярного апарату нирок шляхом відщеплення просегмента за участю ферментів катепсину В або проконвертази 1 [35-37]. Цей процес може відбуватися і в мозковому шарі надниркових залоз [36] (рис. 4).
Утворений ренін зберігається у щільних пухирцях, а його виділенню у кров сприяють: 1) зниження перфузійного тиску в аферентних артеріолах ниркових клубочків (CАТ <90 мм рт. ст.); 2) низька концентрація натрію у фільтраті дистального звивистого канальця нефрона в ділянці, що прилягає до ниркового тільця (macula densa); 3) посилення активності симпатичної нервової системи [38, 37].
На сьогодні ідентифіковано два типи рецепторів, з якими можуть зв’язуватися як проренін, так і ренін, що викликає цілком різні біологічні ефекти. Маннозо‑6-фосфатні рецептори, ідентичні з рецепторами інсуліноподібного фактора росту ІІ, виявлені у кардіоміоцитах, фібробластах, ендотеліоцитах та непосмугованих м’язових клітинах стінок артерій. Зв’язуючись з ними, проренін та ренін депонуються всередині клітин. Це пояснює, чому після білатеральної нефректомії ренін повільніше зникає із судинної стінки, ніж із циркулюючої крові, хоча відомо, що клітини судин не здатні його синтезувати. Отже, цей тип рецепторів забезпечує кліренс прореніну та реніну [39-41].
Власне про(ренінові) білкові рецептори містять 350 амінокислотних залишків та один трансмембранний домен, знайдені у мезангіальних клітинах та клітинах дистальних і збірних канальців нирок, кардіоміоцитах та непосмугованих м’язових клітинах судинної стінки, а також у мозку та судинних структурах і синцитіотрофобластах плаценти [42, 43]. Цей тип рецепторів було відкрито та клоновано групою науковців під керівництвом Женев’єви Нгуєн (Nguyen G.) у 2002 році [42]. При зв’язуванні з ними проренін набуває каталітичної активності без відщеплення просегмента (рис. 4), а ренін – посилює свою ферментативну активність у 4-5 разів. Це сприяє швидкому розщепленню ангіотензиногену з утворенням апгіотензину І на поверхні клітин, де він легко контактує з тканинним АПФ та рецепторами ангіотензину [27, 42].
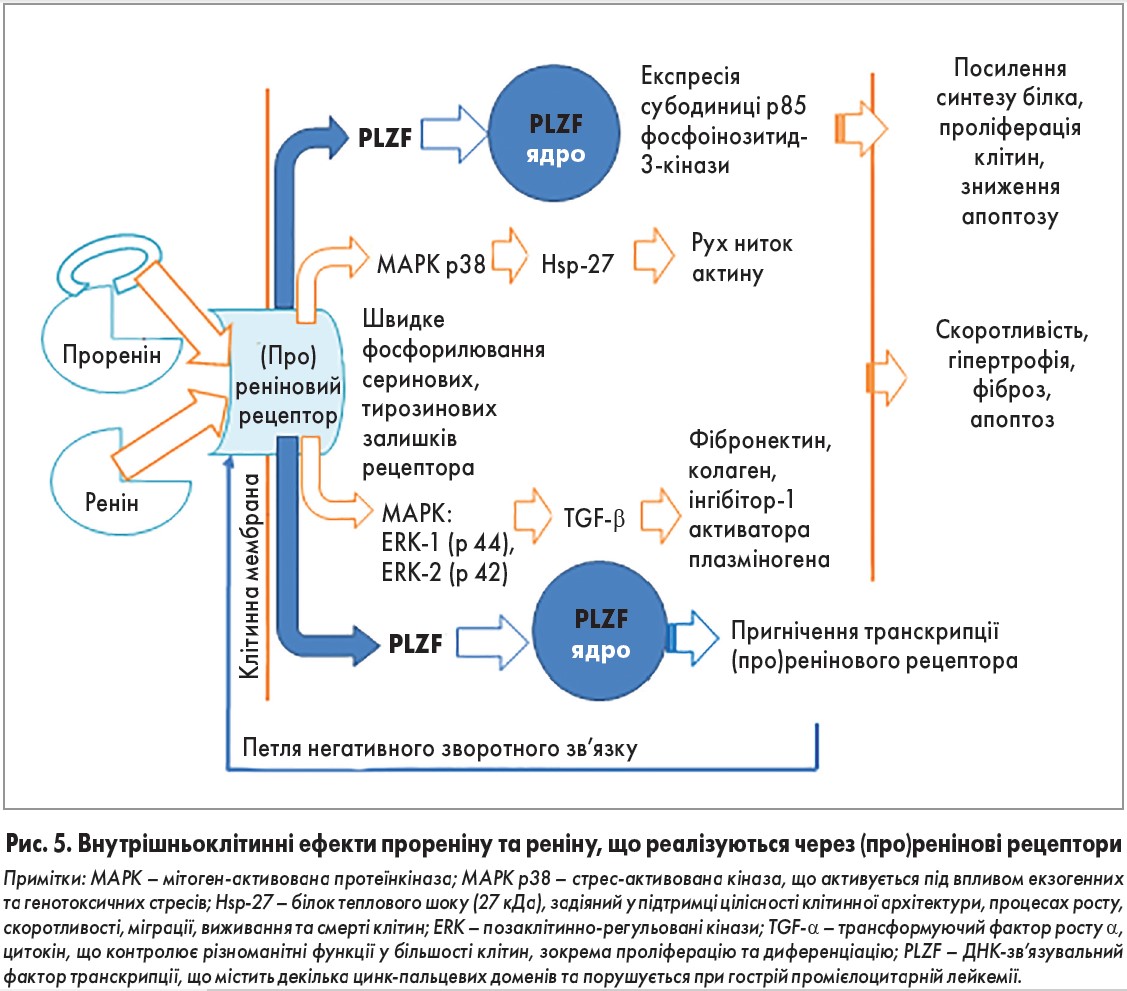
За умов високої експресії рецепторів зв’язування з реніном чи прореніном викликає не утворення ангіотензину, а активацію внутрішньоклітинних сигнальних шляхів мітоген-активованої протеїнкінази, задіяних у процесах проліферації, гіпертрофії, фіброзу та апоптозу (рис. 5). У дослідженнях in vitro встановлено, що для запуску внутрішньоклітинних сигнальних шляхів концентрація про (реніну) у плазмі крові повинна бути на декілька порядків вищою від фізіологічних рівнів. Тому цей процес, очевидно, відбувається лише в органах, здатних синтезувати ренін чи проренін. У мезангіальних клітинах та клітинах збірних трубок канальців нирок це відіграє роль у патогенезі діабетичної нефропатії [44, 45].
Нещодавно з’ясовано, що активація рецептора під впливом реніну стимулює переміщення фактора транскрипції PLZF до ядра, внаслідок чого пригнічується транскрипція самого (про)ренінового рецептора (рис. 5). Це означає, що високий рівень реніну, що виникає при фармакологічній блокаді РААС, пригнічує експресію (про)ренінових рецепторів, у такий спосіб запобігаючи їхній надмірній активації. Чи може проренін викликати аналогічний ефект, поки що невідомо. Натомість при зв’язуванні прореніну з PLZF активується експресія субодиниці p85 фосфоінозитид‑3-кінази, що посилює синтез білка і проліферацію клітин та пригнічує апоптоз [46].
Продовження читайте у Медична газета «Здоров’я України 21 сторіччя» № 21 (442), листопад 2018 р.
Медична газета «Здоров’я України 21 сторіччя» № 20 (441), листопад 2018 р.
СТАТТІ ЗА ТЕМОЮ Кардіологія
Як відомо, кальцій бере участь у низці життєво важливих функцій. Хоча більшість досліджень добавок кальцію фокусувалися переважно на стані кісткової тканини та профілактиці остеопорозу, сприятливий вплив цього мінералу є значно ширшим і включає протидію артеріальній гіпертензії (передусім у осіб молодого віку, вагітних та потомства матерів, які приймали достатню кількість кальцію під час вагітності), профілактику колоректальних аденом, зниження вмісту холестерину тощо (Cormick G., Belizan J.M., 2019)....
Після десятиліть, а часом і запеклих суперечок про переваги та недоліки застосування глюкокортикоїдів (ГК) досягнута певна конвергенція. Сучасні рекомендації лікування таких захворювань, як ревматоїдний артрит (РА), ревматична поліміалгія (РПМ) та васкуліт великих судин відображають поточний стан консенсусу терапії ГК. Однак залишаються відкритими питання щодо можливості тривалого лікування дуже низькими дозами ГК у пацієнтів із РА, а також успішності пошуку інноваційних ГК (лігандів ГК-рецепторів) із покращеним співвідношенням користь/ризик....
Торакалгія – симптом, пов’язаний із захворюваннями хребта. Проте біль у грудній клітці може зустрічатися за багатьох інших захворювань, тому лікарям загальної практики важливо проводити ретельну диференційну діагностику цього патологічного стану та своєчасно визначати, в яких випадках торакалгії необхідна консультація невролога. В березні відбувся семінар «Академія сімейного лікаря. Біль у грудній клітці. Алгоритм дій сімейного лікаря та перенаправлення до профільного спеціаліста». Слово мала завідувачка кафедри неврології Харківського національного медичного університету, доктор медичних наук, професор Олена Леонідівна Товажнянська з доповіддю «Торакалгія. Коли потрібен невролог»....
Рівень ліпопротеїну (a) >50 мг/дл спостерігається в ≈20-25% населення і пов’язаний із підвищеним ризиком серцево-судинних захворювань (ССЗ) [1]. Ліпопротеїн (a) задіяний в атерогенезі та судинному запаленні, а також може відігравати певну роль у тромбозі через антифібринолітичну дію і взаємодію із тромбоцитами [2, 3]. Дієта та фізична активність не впливають на рівень ліпопротеїну (a); специфічної терапії для його зниження також не існує. Підвищений ризик ССЗ, пов’язаний з ліпопротеїном (а), залишається навіть у пацієнтів, які приймають статини [4]. Саме тому існує критична потреба в терапії для зниження цього ризику, особливо в первинній профілактиці. ...