



Лабораторная диагностика тромботической микроангиопатии
Тромботическая микроангиопатия (ТМА) – это патологический процесс, сопровождающийся системным тромбозом вследствие окклюзии сосудов микроциркуляторного русла (артериол, капилляров) различных органов, который приводит к микроангиопатической гемолитической анемии (МАГА), тромбоцитопении и повреждению органов [1-3].
ТМА классифицируют на первичные и вторичные. Первичные ТМА включают в себя тромботическую тромбоцитопеническую пурпуру (ТТП), типичный гемолитико-уремический синдром (тГУС) и атипичный (аГУС). К многочисленным вторичным ТМА относятся: преэклампсия, эклампсия, HELLP‑синдром, аутоиммунные заболевания, злокачественные опухоли, вирус иммунодефицита человека, грипп H1N1, гломерулопатии, метилмалоновая ацидурия с гомоцистинурией, некоторые лекарственные препараты, ионизирующее излучение, трансплантация солидных органов и костного мозга.
Наиболее известными заболеваниями, в основе которых лежит ТМА, являются тромботическая тромбоцитопеническая пурпура и гемолитико-уремический синдром.
Тромботическая тромбоцитопеническая пурпура
ТТП является редким гематологическим заболеванием с распространенностью 10 случаев на 1 млн человек. Первый острый эпизод возникает в зрелом возрасте (90% всех случаев ТТП), но может встречаться у детей и подростков (10%). Кроме аутоиммунного механизма, лежащего в основе ТТП, описаны редкие неиммунные формы (синдром Апшоу-Шульмана). ТТП встречается в 2 раза чаще у женщин и ее клиническое течение характеризуется тенденцией к рецидивам. Несмотря на терапевтическое лечение, ТТП остается опасным для жизни заболеванием с уровнем смертности от 10 до 20%.
Патофизиология ТТП
Начиная с 1924 г. (Эли Мошковиц описал фатальный клинический случай тромботической микроангиопатии) и до 80-90-х годов XX ст., этиология ТТП оставалась неизвестной со смертельным исходом в 90% случаев. Эмпирическое лечение с заменой плазмы у пациентов в этот период показало значительное улучшение прогноза при ТТП, что позволило добиться выживаемости в 85% случаев.
Роль фактора Виллебранда и дефицит ADAMTS13 в развитии ТТП
Фактор фон Виллебранда является мультимерным белком, участвующим в инициации слипания тромбоцитов. Он синтезируется и высвобождается эндотелиальными клетками и хранится в органеллах цитоплазмы, называемыми гранулами Вайбеля-Паллада. Мономеры фактора Виллебранда с м. в. 200-300 кДа образуют мультимеры глобулярной конформации с м. в. 500-30 000 кДа, которые снижают его взаимодействие с тромбоцитами.
Связь между патофизиологией ТТП и фВ была показана в работе J. Moake. Он обнаружил, что у пациентов с хроническим рецидивирующим ТТП в больших количествах циркулирует высокомолекулярный фВ – как в период острой фазы, так и во время ремиссии. Так как эти необычно большие мультимеры отсутствуют в нормальной плазме, исследователь предположил, что должна существовать протеаза для расщепления гиперадгезивных мультимеров фВ. Таким белком оказался ADAMTS13 (A Disintegrin And Metalloprotease with ThromboSpondin‑1-like domains, member 13) – металлопротеаза, принадлежащая семейству пептидазных белков, которые в 1996 г. независимо друг от друга выделили исследователи H. M. Tsai и M. Furlan. ADAMTS13 синтезируется в звездчатых клетках печени и эндотелиальных клетках сосудов в виде гликопротеина с м. в. 180 кДа [22, 23]. Кроме того, биологически активный ADAMTS13 синтезируют подоциты почек, тубулярные эпителиальные клетки и тромбоциты [24-26]. После структурных изменений в эндоплазматическом ретикулуме ADAMTS13 становится активным и выделяется в качестве активного фермента в циркуляцию крови [27], где происходит расщепление высокомолекулярного фВ между остатками тирозина 842 и метионина 843 домена А2. При дефиците ADAMTS13 наблюдается фВ‑зависимое накопление тромбоцитов, что в конечном итоге приводит к микрососудистому тромбозу и ТТП (рис. 1).
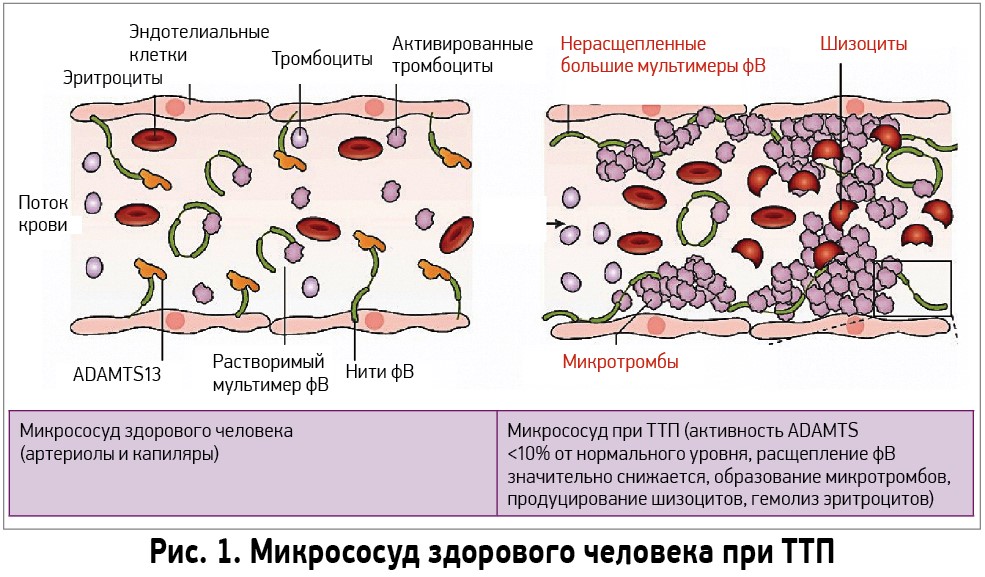
ADAMTS13 регулирует функциональную активность фВ, способствуя ограничению роста тромбов в микроциркуляторном русле. Дефицит ADAMTS13 может быть обусловлен мутациями генов, которые кодируют синтез этого фермента, или выработкой к нему антител.
Лабораторная диагностика ТТП
Диагностика ТТП с участием ADAMTS13 включает определение следующих лабораторных тестов [28]:
- активность ADAMTS13 для подтверждения клинического диагноза ТТП;
- анти-ADAMTS13 IgG для установления причины дефицита ADAMTS13;
- в отдельных случаях – секвенирование гена ADAMTS13.
Для дифференциальной диагностики приобретенной и врожденной формы ADAMTS13 применяют алгоритм.
Уровни ADAMTS13 <10% при наличии антител характерны для большинства пациентов с ТТП. Эти пациенты реагируют на обмен плазмы и иммуносупрессию. Пациенты с уровнями ADAMTS ≥10% и без антител не будут реагировать на такие методы лечения. Для них необходимо искать другие причины тромбоцитопении и анемии, которые включают: ДВС‑синдром, сепсис, рак, преэклампсию, системный склероз, СКВ, злокачественную гипертензию, отторжение почечного трансплантанта. Среди пациентов редко встречается наличие низкого уровня ADAMTS13 без присутствия аутоантител. Такие пациенты должны пройти генетическое тестирование ADAMTS13, так как им потребуется только инфузия плазмы без иммуносупресси. Генетическое тестирование также показано для детей, женщин с рецидивирующими эпизодами во время беременности, людей с положительным семейным анамнезом или при наличии других клинических признаков.
Интерпретация теста ADAMTS13
Норма: Активность – 50-140%, ингибитор не выявлен.
- Активность – 0-5%, острая фаза синдрома Мошковица. Пациенты нуждаются в немедленном введении плазмы с металлопротеиназой.
- Активность – 6-7%, признак генетической мутации и синдрома Апшоу-Шульмана.
- Активность – ниже 40%, осложнения беременности, ДВС‑синдром, онкологические заболевания, послеоперационные вмешательства.
Гемолитико-уремический синдром
ТМА с первичным обнаружением почечной недостаточности называется гемолитическим уремическим синдромом (ГУС).
Наиболее распространенной формой является заражение кишечной палочкой Escherichia coli (серотипы O157:H7, O111:H8, O103:H2, O123, O26 или др., которые продуцируют шигатоксин), что сопровождается кровавой диареей. Эта форма называется EHEC-ГУС (энтерогеморрагическая E. coli, EHEC) или STEC-ГУС [29]. ГУС при респираторной инфекции (Streptococcus pneumoniae) или SP-ГУС встречается крайне редко [30]. Если ни одна из инфекций не определена, тогда подозревают атипичный ГУС, который обусловлен генетическими нарушениями или изменениями иммунной системы, характеризующимися патологией системы комплемента [31]. У детей около 80-90% случаев вызваны EHEC-ГУС, 5-10% относятся к аГУС и менее 5% – к SP-ГУС [32].
Все патофизиологические формы ГУС имеют опосредованное повреждение эндотелиальных клеток, которое в основном поражает капилляры почек. Если ГУС обусловлен действием кратковременного триггера (запускающий фактор), такого как шигатоксин (энтерогеморрагическая Escherichia coli), инфекции Streptococcus pneumoniae или лекарственных препаратов, то после удаления триггера и поддерживающей терапии происходит спонтанная ремиссия. В случае генетического дефекта или приобретенной дисрегуляции комплемента или системы гемостаза ТМА может привести к повреждению органов (обычно почки) даже после удаления триггера. Примером этого являются комплемент-регуляторные дефекты, вызванные мутациями фактора Н, фактора I, факторов В, С3 или мембранным кофактором белка (МСР) или аутоантителами к фактору Н. Запускающими факторами также могут быть: злокачественная опухоль, беременность, трансплантация стволовых клеток, инфекции, лекарственные препараты. В редких случаях наблюдаются мутации генов, участвующих в системе свертывания крови, такие как диацилглицеролкиназа E (DGKE) и тромбомодулин, которые характерны в основном для детей до одного года [33].
Типичный гемолитико-уремический синдром
Типичный гемолитико-уремический синдром (тГУС)– это острое заболевание, при котором на фоне инфекционно-обусловленной диареи в продромальном периоде развиваются неиммунная микроангиопатическая гемолитическая анемия, тромбоцитопения и острая почечная недостаточность.
В 1955 г. С. Gasser ввел термин у пациентов с почечной недостаточностью после кровавой диареи, а в 1978 г. M. Rahaman показал связь между ГУС и Shigella инфекцией [34]. Самым распространенными продуцентами шигатоксина являются серотипы EHECO157:H7 и O104:H4.
Основным фактором патогенности считается шигаподобный токсин (Stx), который продуцирует также Shigella dysenteriae 1 типа. Выделяют два семейства Stx – Stx1 и Stx2. Показано, что E. сoli, выделенная от больных ГУС, продуцирует либо оба токсина (Stx1 и Stx2), либо только Stx2. Источниками передачи шигатоксина являются: крупный рогатый скот, олени, овцы, козы, лошади, собаки, птицы, мухи. А источники заражения для человека: навоз и кормушки в хозяйствах (повышенный риск заражения в сельской местности); молоко; мясо (при убое скота); нехлорированная вода; грязные фрукты и овощи (ростки редиса, салат, яблочный сидр); контакт с инфицированными животными или экскрементами человека; непастеризованный яблочный сок; контакт с зараженными людьми (в детских учреждениях). Средний интервал проявления болезни составляет 3 дня (от 1 до 8 дней). Типичными начальными проявлениями являются: спазмы в брюшной полости, слабость, олигоанурия. Геморрагическая диарея встречается в 70% случаев. ГУС обычно диагностируется через 6 дней после начала диареи с такими симптомами как рвота (30-60%), лихорадка (30%), повышение количества лейкоцитов.
Патофизиологический механизм тГУС
При кишечной инфекции шигатоксин проходит через стенку кишечника и транспортируется в кровоток нейтрофилами, моноцитами и тромбоцитами. Далее связывается с эндотелиальными клетками почки через CD77 (кластер дифференцировки, CD77 или глоболтриаосилцерамид, Gb3), вызывая гибель клеток и высвобождая фактор Виллебранда. Интерлейкины (IL‑1, IL‑6) и фактор некроза опухоли (TNF) усиливают экспрессию рецепторов шигатоксинов на поверхности эндотелиальных клеток. Поврежденные клетки экспрессируют поверхностный высокомолекулярный фактор Виллебранда, который инициирует слипание тромбоцитов через взаимодействие с гликопротеином Ib. Шигатоксины также вызывают экспрессию тканевого фактора на эндотелиальных клетках, что приводит к активации фактора VII и образованию фибрина. При этом происходит тромбоз почечных сосудов, который может захватывать другие сосуды. Наблюдается активация системы комплемента и дальнейшее разрушение эндотелиальных клеток.
Лабораторная диагностика ГУС
Лабораторные исследования следует выполнять в первые сутки госпитализации пациента в стационар до начала антибактериальной терапии. При этом показаны следующие тесты:
- посев кала для выявления культуры STEC (cреда MacConkey для E. сoli O157:H7);
- определение в сыворотке крови антител к липополисахариду (O157 LPS);
- определение шигатоксина в кале или ректальном мазке методом ПЦР;
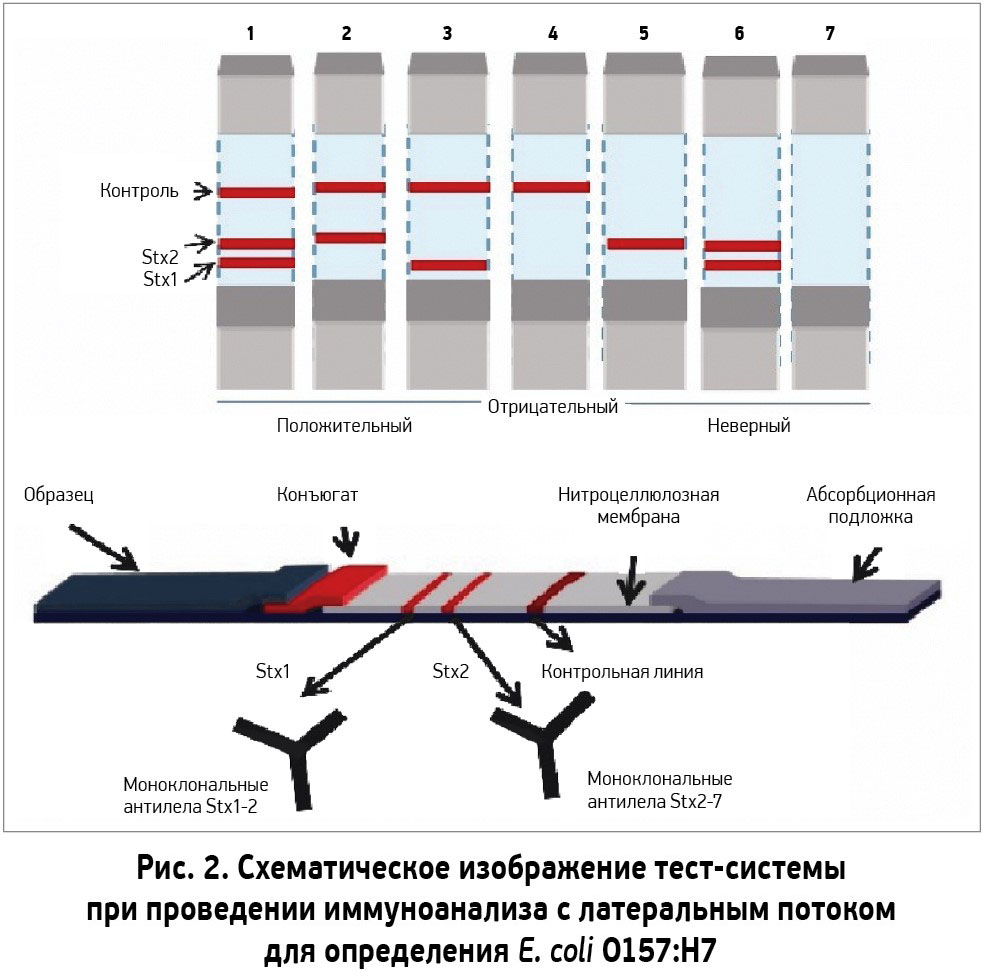
- разработка быстрых тестов для определения E. сoli O157:H7 в кале [35] (рис. 2).
Атипичный гемолититико-уремический синдром
Атипичный гемолитико-уремический синдром (аГУС) – хроническое системное заболевание генетической природы, в основе которого лежит неконтролируемая активация альтернативного пути комплемента, ведущая к генерализованному тромбообразованию в сосудах микроциркуляторного русла (комплемент-опосредованная тромботическая микроангиопатия). В 1998 г. Р. Warwicker и соавт. первыми связали развитие аГУС с генетическими нарушениями, опубликовав наблюдение развития аГУС.
Распространенность аГУС составляет 10% от распространенности типичного ГУС (0,2 на 100 тыс. населения, из которых 40% больных аГУС – взрослые). Атипичный ГУС подразделяют на семейный (10-20%) и спорадический (80-90%). В основе аГУС лежат мутации регуляторных белков системы комплемента, такие как: комплементарный фактор H (CFH), мембранный кофакторный протеин (МСР), комплементарный фактор I (CFI), тромбомодулин (THBD), фактор (В), фактор С3, а также антитела к CFH.
Патофизиологический механизм аГУС
При активации комплемента образуется C3-конвертаза, расщепляющая C3 на малый (С3а) и большой (C3b) фрагменты, которая опсонизируется на поверхности микробной клетки и формирует мембраноатакующий комплекс (МАК), состоящий из C5b, C6, C7, C8 и C9. Это приводит к осмотическому лизису клетки. Чтобы активированная система комплемента не уничтожила собственные клетки, на их поверхности расположены белки-регуляторы. Часть таких белков синтезируется в печени и циркулирует в плазме крови в неактивном состоянии. К таким белкам относят комплементарный фактор H (CFH), фактор I (CFI) и мембранный кофакторный протеин, закрепленный на поверхности клеток (CD46). Фактор I, главный из вышеперечисленных факторов, расщепляет C3b и C4b. Фактор Н и CD46 являются кофакторами фактора комплемента I. Первый из них связывается с гликозаминогликанами собственных клеток организма, отсутствующими на мембранах бактериальных клеток, а также ингибирует активность C3-конвертазы. При мутации данных регуляторных белков происходит утрата защиты эндотелиальных клеток от повреждения конечными продуктами активации альтернативного пути комплемента (рис. 3).
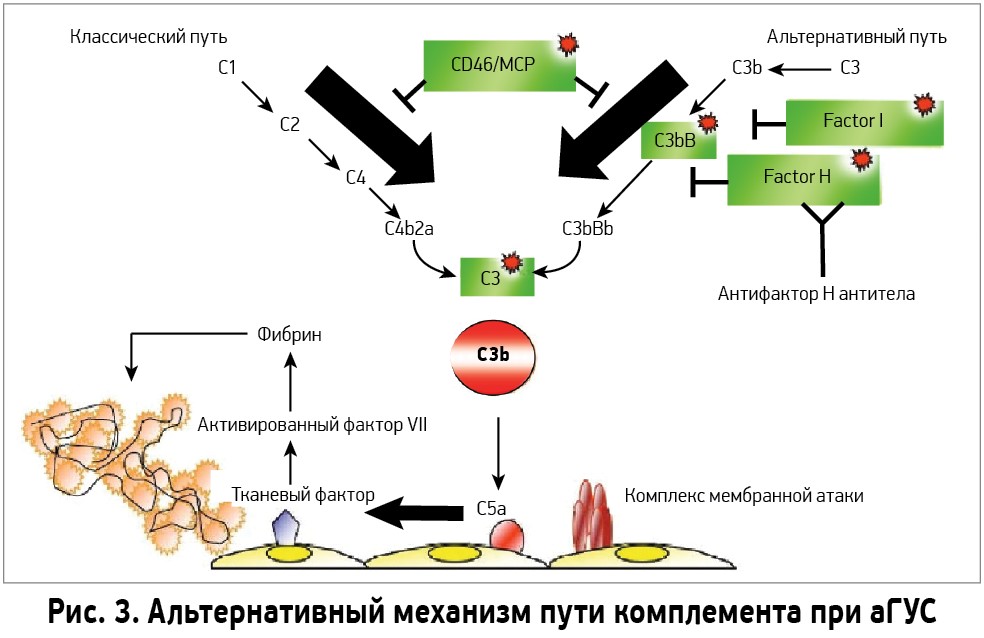
Мутации в генах, имеющиеся у пациентов с аГУС, приводят к нарушению защиты эндотелиальных клеток (ЭК) от активации системы комплемента вследствие дефицита или функциональных нарушений белков-регуляторов. Это вызывает повреждение ЭК с последующим образованием тромбов в сосудах микроциркуляторного русла. Преимущественное поражение почек при аГУС связано с особым строением эндотелия клубочков, которое обеспечивает повышенную чувствительность к повреждению, обусловленному нарушенной регуляцией комплемента.
Наиболее частой причиной аГУС считается мутация фактора Н – основного регуляторного белка, ограничивающего активность альтернативного пути комплемента. Его дефицит, как и дефицит других регуляторных протеинов, как плазменных, так и мембраносвязанных (факторы: Н, I, MCP), приводит к безудержной и постоянной активации альтернативного пути, завершающегося образованием мембраноатакующего комплекса. Последний поддерживает механизмы сосудистого повреждения и тромбообразования. У небольшого числа больных спорадическая форма аГУС может быть обусловлена присутствием антител к фактору Н.
На сегодняшний день аГУС рассматривают как катастрофически протекающее угрожающее жизни системное заболевание с неблагоприятным прогнозом (70% больных умирают в момент первого эпизода).
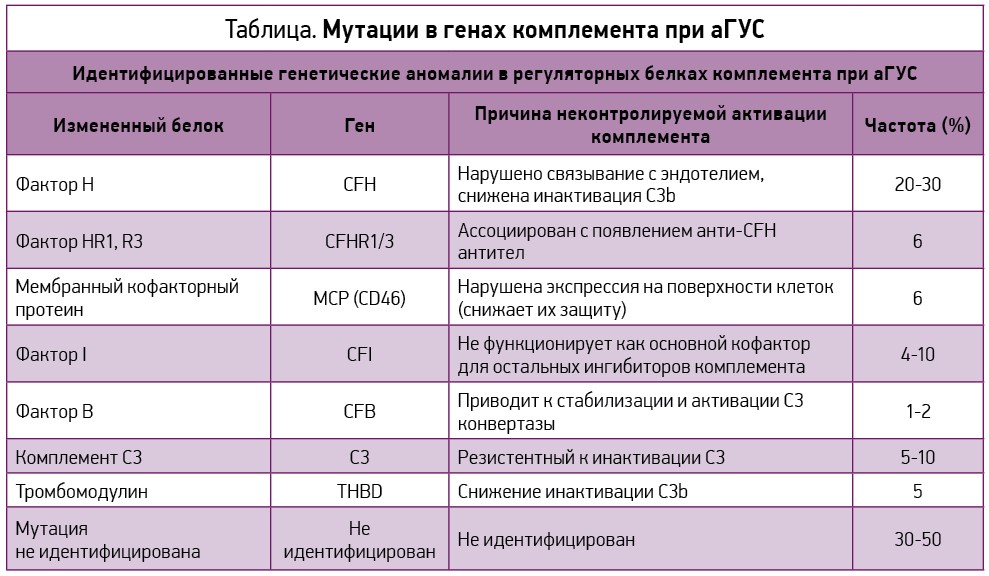
Мутации в генах комплемента при аГУС представлены в таблице [36].
Лабораторные тесты при аГУС
Для подтверждения аГУС рекомендовано проведение следующих тестов [37]:
- мутации/делеции генов комплемента (С3, CD46 (MCP), CFB, CFH, CFHR1, CFHR3, CFHR4, CFHR5, CFHI);
- мутации генов белков системы коагуляции (плазминоген, тромбомодулин);
- анти-CFH.
Выводы
ТМА может проявляться при самых разных заболеваниях и относится к неотложным гематологическим заболеваниям, которые требуют срочного вмешательства. В настоящее время для таких форм ТМА, как ТТП и ГУС, определены молекулярные механизмы их развития и ранние методы лабораторной диагностики. Также имеется возможность дифференцировать ТТП от аГУС. Так, для ТТП характерен дефицит ADAMTS13, при котором мультимеры фактора Виллебранда не расщепляются. Данный лабораторный тест позволит врачам отличить ТТП от аГУС (включая исследование мутации генов комплемента). К сожалению, специфические методы диагностки, такие как определение активности ADAMTS13, анализ на шигатоксин, не только не входят в перечень исследований экспресс-лабораторий, но они доступны не всем крупным медицинским учреждениям. Однако рутинные тесты, выполняемые во многих лабораториях (повышение ЛДГ, снижение гаптоглобина, идентификация шизоцитов, отрицательный тест Кумбса), за очень короткий промежуток времени позволят подтвердить соответствующий диагноз.
Список литературы находится в редакции.
Медична газета «Здоров’я України 21 сторіччя» № 21 (466), листопад 2019 р.
СТАТТІ ЗА ТЕМОЮ Діагностика
Еритроцити, або червоні кров’яні тільця, є найпоширенішим компонентом клітин крові, які становлять 40-45% їхнього обсягу. Плазматична мембрана еритроцитів має унікальну будову, що надає їм біологічних і механічних властивостей, необхідних для виконання специфічних функцій. Основна функція еритроцитів в організмі – це транспорт кисню, опосередкований гемоглобіном. Вони активно беруть участь як в артеріальних, так і у венозних тромбозах [1]. Гемоглобін – висококонсервативний білок, який завдяки своїй здатності зворотно зв’язувати кисень бере участь у процесах, що лежать в основі аеробного життя на планеті Земля. Головна роль цього білка полягає у підтримці клітинного гомеостазу. Однак завдяки майже 200-річним дослідженням гемоглобіну тепер відомо, що цей білок також відіграє важливу роль в інших метаболічних процесах, як-от передача сигналів у клітинах, модуляція запальної реакції, за тромбозу при гемолізі еритроцитів тощо [2-4]....
У сучасному світі онкологічні захворювання становлять чи не найбільшу загрозу життю людини, поступаючись тільки серцево-судинній патології [1]. Більшість неінфекційних хвороб людини, зокрема й онкологічні захворювання, є багатофакторними, і їх розвиток пов’язаний у тому числі з генетичними чинниками. Це, з одного боку, підвищує з віком ризик виникнення хронічних захворювань, а з іншого – дає змогу разом з лікарем розробити заходи з їх профілактики, раннього виявлення й ефективного лікування [2]....
Власна патоморфологічна лабораторія – необхідність для всіх клінік ендоскопічного, хірургічного й онкологічного профілю. Одним із видів діяльності такої лабораторії є проведення інтраопераційних досліджень. Ці дослідження виконують для визначення тактики подальшого оперативного втручання під час операції. Тому створення лабораторії на колесах, що може надавати результати патоморфологічного дослідження в будь-якому місці та ще під час операційного втручання, стало логічним рішенням для Медичної лабораторії CSD LAB, найбільшої патоморфологічної лабораторії України та Східної Європи....
Нефракціоновані (НФГ) і низькомолекулярні гепарини (НМГ) є препаратами, що широко використовуються та запобігають артеріальним і венозним тромбозам. Однак їхнє застосування також пов’язано з парадоксальною реакцією, що зумовлює потенційно небезпечний для життя протромботичний стан, результатом чого є серйозні ускладнення (гангрена, ампутація кінцівок) або фатальні наслідки. Гепарин-індукована тромбоцитопенія (ГІТ) – це імуноопосередкована відповідь на введення гепарину, який спричиняє небезпечний для життя тромбоз і є клінічно значущим негеморагічним ускладненням. ГІТ вважається потенційно загрозливим для життя станом за терапії гепарином, що спричиняє утворення нових згустків крові, а не сприяє запобіганню утворення нових тромбів. Хоча при введенні гепарину імунна реакція зустрічається досить часто (від 8 до 50%), клінічні ускладнення у разі ГІТ виникають у ≈0,2-3% пацієнтів, які приймають гепарин протягом >4 дні; частіше спостерігаються в жінок [1-3]. У цьому стані тромбоцити різко знижуються до рівня ≥20×109/л. Смертність становить 10-20%....