



Хвороба Фабрі під масками гіпертрофії міокарда, кардіоміопатії та ниркової недостатності
У межах онлайн-вебінару «Орфанні захворювання. Хвороба Фабрі – як надати шанс пацієнту», організованого за підтримки ТОВ «Санофі-Авентіс Україна», доступно й ґрунтовно було надано сучасну інформацію про основні клінічні прояви та детально розкрите питання забезпечення діагностики й лікування хвороби Фабрі (ХФ) із погляду кардіолога. Своєю думкою із цього приводу поділився завідувач відділу ішемічної хвороби серця і метаболічних порушень ДУ «Національний інститут терапії iменi Л.Т. Малої НАМН України» (м. Харків), старший науковий співробітник, д. мед. н. Сергій Андрійович Серік.
С.А. Серік у своїй доповіді приділив увагу хворобі накопичення глікосфінголіпідів, що вперше було описано 1898 р. Йоганесеном Фабрі (Німеччина) та Вільямом Андерсоном (Велика Британія) незалежно один від одного (Desnick et al., 2001). Виникнення захворювання зумовлене генною мутацією на довгому плечі Х-хромосоми, в локусі Xq22.1, що викликає дефіцит α-галактозидази A (α-GLA) і накопичення субстрату – нейтральних глікосфінголіпідів, головним чином – глоботріаозилцерамідів (GL‑3). Тип успадкування – Х-зчеплений рецисивний із неповною пенетрантністю у жінок, для чоловіків же характерна класична форма патології.
Жіночий фенотип ХФ із безсимптомним або легким перебігом пояснюється феноменом інактивації Х-хромосом (лайонізацією) – процесом, у ході якого інактивується одна з двох копій X-хромосом, представлених у клітинах. Кожен орган в організмі жінки має свій власний патерн інактивації Х-хромосоми. У жінки з дефектним геном в одному органі здатні експресуватися 60% її здорових Х-хромосом, тоді як в іншому – працювати тільки на 30%. Симптоми ХФ можуть мати місце у 70% гетерозиготних жінок. Іноді прояви бувають такими ж тяжкими, як у чоловіків (Pinto et al., 2010).
За даними реєстру пацієнтів із ХФ у США, поширеність хвороби становить від 1:40 тис. до 1:60 тис. серед чоловіків та 1:20 тис. – серед жінок. Цей показник варіює у різних країнах. Наприклад, в Австралії поширеність ХФ у загальній популяції складає 1:117 тис., у Норвегії з-поміж чоловіків – 1:17 тис. За деякими даними частота серед чоловіків може досягати 1:1600 – 1:3100. Так, результати скринінгу понад 110 тис. новонароджених у Тайвані виявили зниження активності α-GAL та характерні мутації з частотою 1:2400. У хлопчиків частота мутації IVS4+919G^А, що викликає кардіальний варіант ХФ, була 1:1600. Подібний скринінг близько 37 тис. новонароджених хлопчиків провели в Італії, частота дефіциту α-GAL досягала 1:3100, у більшості випадків (11:1) відзначалися мутації, асоційовані з пізнім, а не «класичним» (дебют у дитячому віці) фенотипом захворювання (Germain, 2010; Hsu, Niu, 2017; Wasserstein et al., 2018; Colon et al., 2017; Lu et al., 2018).
Патогенез ХФ полягає у прояві мутації у вигляді дефіциту GLA, що призводить до накопичення GL‑3 у різних функціональних клітинах органів, як-то ендотеліоцити (кровоносні судини), подоцити (нирки), нейрони (ганглії вегетативної нервової системи), кардіоміоцити, провідна система (серце), потові залози (шкіра) (Masson et al., 2004).
Клінічні прояви ХФ, за словами С.А. Серіка, включають:
1. Кардіальні симптоми (Schiffmann et al., 2009; Kampmann et al., 2002; Linhart et al., 2002):
- аритмії (фібриляція передсердь, шлуночкові тахікардії);
- атріовентрикулярні (АВ) блокади;
- вкорочення інтервалу P-R;
- дисфункція клапанів (недостатність мітрального й аортального);
- гіпертрофія міокарда (ГМ) лівого шлуночка (ЛШ);
- зниження толерантності до фізичного навантаження;
- розширення кореня аорти;
- стенокардія, інфаркт міокарда (ІМ);
- серцева недостатність (СН);
- раптова смерть.
2. Нефрологічні ускладнення:
- протеїнурія;
- зниження швидкості клубочкової фільтрації (ШКФ);
- хронічна ниркова недостатність.
3. Периферична невропатія, що проявляється:
- хронічним болем;
- больовими кризами;
- парестезіями;
- порушенням чутливості.
4. Порушення моторики шлунково-кишкового тракту (ШКТ), що супроводжується:
- спастичним болем;
- діареєю;
- метеоризмом;
- нудотою.
5. Дерматологічні прояви (Eng et al., 2006):
- ангіокератома;
- гіпогідроз (сухість шкіри, непереносимість спеки, холоду та фізичних навантажень).
Так, типовим вважається невропатичний біль у кінцівках (акропарестезії), що є проявом невропатії дрібних волокон Фабрі та, відповідно, відображенням дисфункції нейронів маломієлінізованих і немієлінізованих нервових волокон. Цей пекучий або інтенсивний біль у кистях, стопах виникає у відповідь на невелике больове подразнення чи підвищення температури навколишнього середовища, особливо як реакція на теплу і гарячу воду. Може супроводжуватись парестезіями – болісними відчуттями поколювання чи повзання мурашок у кінцівках (Tavee et al., 2009).
Крім постійного, хронічного невропатичного болю і акропарестезій для ХФ характерні больові кризи. Кризи Фабрі виникають епізодично при зміні погоди, лихоманці, фізичному навантаженні, стресі й після приймання алкоголю. Біль стає інтенсивнішим і набуває нестерпного характеру. Може тривати від декількох хвилин до декількох тижнів та не минає на тлі введення наркотичних анальгетиків. Кризи часто супроводжуються субфебрильною лихоманкою та підвищенням швидкості осідання еритроцитів, що нерідко призводить до встановлення помилкових діагнозів: ревматоїдного артриту, ревматичної лихоманки, артритів, еритроміалгії, синдрому Рейно або «хвороби росту».
Дисфункція вегетативної нервової системи, що також є проявом невропатії тонких волокон, призводить до (Cable et al., 1982):
- порушення моторики ШКТ (постпрандіальний спастичний біль та здуття живота, діарея, що виникає періодично, відчуття швидкого насичення, нудота, блювання, втрата маси тіла);
- гіпогідроз, ангідроз;
- порушення продукції слини;
- порушення цереброваскулярного кровотоку;
- зниження ортостатичного серцево-судинного контролю.
Ангіокератоми – червонуваті, з фіолетовим відтінком точкові висипання на шкірі до 5 мм у діаметрі, що не бліднуть при натисканні, розташовуються від пупка до області стегна, зустрічаються у 66% пацієнтів чоловічої статі та у 36% жінок із ХФ. Окрім того, ймовірна поява телеангіоектазій, які розташовуються на ділянках шкіри, що зазнають сонячного впливу – обличчі та шиї.
Специфічним симптомом ХФ також є помутніння рогівки (воронкоподібна кератопатія). Можливі формування задньої субкапсулярної катаракти й ураження судин сітківки та рогівки.
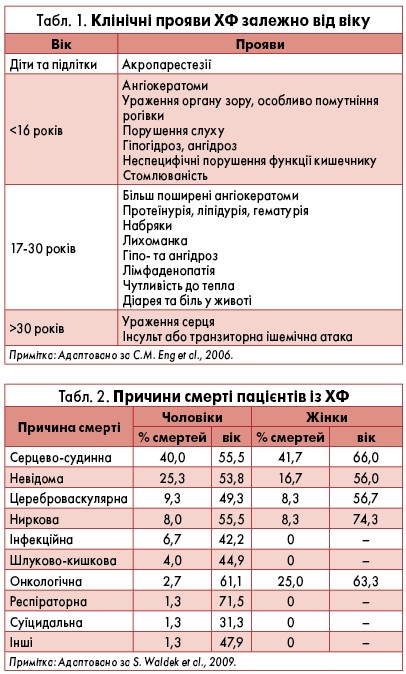
Оскільки на кожному етапі життя пацієнта клінічні прояви ХФ мають специфічні особливості, спікер надав інформацію про вікові відмінності симптомів (табл. 1).
Прогресування ХФ є наслідком поступового накопичення GL‑3. На початковому етапі, коли відбувається дебют ранніх симптомів (до 20 років), воно є незначним, однак у віці 20‑40 років виразність клінічних проявів наростає, відображає незворотне поліорганне ураження відкладеним GL‑3 та як кінцевий наслідок – передчасну смерть у віці 45‑60 років (Eng et al., 2006).
У дослідженні S. Waldek et al. (2009) у 2848 хворих на ХФ із 39 країн було проаналізовано тривалість життя згідно зі статевою ознакою. Виявлено, що захворювання скорочує життя на 15 років у чоловіків та на 5 років у жінок, а кількість смертей починає зростати з 35 та 50 років відповідно. Головною причиною смерті (40%) як у чоловіків, так і жінок є ураження серця, менш ніж 10% у структурі смертності займають цереброваскулярні ускладнення і ниркова недостатність (табл. 2).
 Як зауважив С.А. Серік, виділяють дві форми ХФ:
Як зауважив С.А. Серік, виділяють дві форми ХФ:
1. Класична: низька або нульова активність α-GAL <1% від норми; характеризується дебютом у дитячому або підлітковому віці та поліорганним ураженням.
2. Атипова: залишкова активність α-GAL (2‑30% від норми), GL‑3 може накопичуватись у капілярах дрібних кровоносних судин; характеризується пізнім дебютом з ізольованим ураженням одного органу – нирок, головного мозку, серця.
При нирковій формі ХФ накопичення GL‑3 в ендотелії судин, подоцитах, мезангіальних клітинах призводить до прогресуючого гломерулосклерозу, тубулярної атрофії, інтерстиціального фіброзу. Перші симптоми нефропатії Фабрі – мікроальбумінурія, протеїнурія. Пізніше починається наростаюче зниження швидкості клубочкової фільтрації та розвивається термінальна ниркова недостатність (Thuberg et al., 2002).
Що стосується уражень ЦНС, то для ХФ властивий високий ризик розвитку транзиторних ішемічних атак, ішемічних та геморагічних інсультів. У 50% перенесений інсульт є дебютом ХФ. У більшості хворих інсульт розвивається у віці від 20 до 50 років, зокрема у кожного п’ятого з них – до 30 років. Частота інсульту складає 6,9% у чоловіків і 4,3% у жінок. Геморагічний інсульт у чоловіків зустрічається частіше, ніж у жінок. У 2/3 пацієнтів інсульт є наслідком атипового перебігу хвороби з ізольованим ураженням головного мозку.
Одним із фатальних наслідків прогресування ХФ є ураження серця, що може бути її єдиним проявом. Патофізіологія у даному випадку полягає в накопиченні GL‑3 у кардіоміоцитах, ендотелії судин, фібробластах клапанів, провідній тканині, що призводить до ГМЛШ, апоптозу/некрозу, запалення, міокардіального фіброзу, ішемії міокарда з розвитком діастолічної, систолічної дисфункції, скорочення інтервалу PQ, аритмій, хронотропної неспроможності та проявляється насамкінець СН, стенокардією, гострим коронарним синдромом, синкопе та раптовою смертю (Hagege et al., 2019).
Patel et al. (2015) вивчали кардіальні події у 207 пацієнтів (47% чоловіків, середній вік – 44 роки) із ХФ протягом семи років. У 58 хворих (28%) виявлено мутації, асоційовані переважно з кардіальним фенотипом. У 21 пацієнта (10%) розвинулася виразна СН (≥3 функціонального класу за класифікацією Нью-Йоркської асоціації серця), у 13 (6%) – фібриляція передсердь, у 13 (6%) було встановлено штучний водій ритму для корекції брадикардії, семеро (3%) померли внаслідок кардіологічної патології. Розвиток кардіальних подій не асоціювався з наявністю кардіогенетичних варіантів – первинний (комбінована точка) та вторинний результати були однаковими в осіб із кардіальним та класичним генетичними варіантами.
Нещодавній систематичний огляд 13 досліджень за участю 4185 пацієнтів із ХФ за період 1,2-10 років свідчить, що 75% від загальної кількості смертей (8,3%) є наслідком кардіальної патології, 62% від усіх летальних випадків асоційовані з раптовою серцевою смертю. Факторами ризику раптової смерті були вік, чоловіча стать, ГМЛШ, нестійка шлуночкова тахікардія (Baig et al., 2018).
Аналіз даних реєстру ХФ продемонстрував ураження серця у 13,9% жінок та 19,3% чоловіків. Серед симптомів найчастіше зустрічалися ГМЛШ та аритмії (табл. 3). Середній вік 1-го клінічного прояву становив 42 роки у чоловіків і 48 років у жінок.
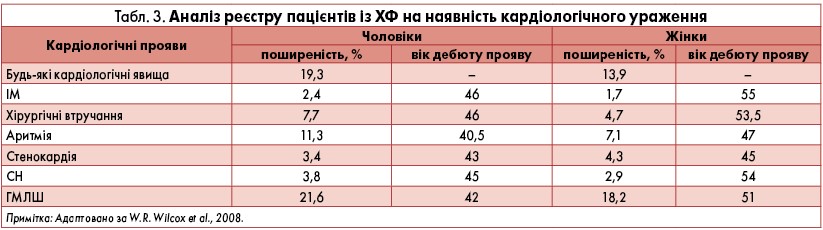
Відповідно до результатів аналізу іншого реєстру – Fabry Outcome Survey – в країнах Західної Європи ГМЛШ за даними ехокардіографії виявляли у 53% чоловіків і 33% жінок із ХФ. Розвиток клінічних проявів ураження серця при ХФ корелював із наявністю ГМЛШ (Linhart et al., 2007). За результатами обстеження 714 пацієнтів із ХФ встановлено, що:
- будь-який кардіальний симптом виявлявся у 89,9% у пацієнтів із ГМЛШ і 50,8% – без ГМЛШ;
- задишка/СН – у 58,8 та 19,9% відповідно;
- стенокардія – у 49,6 та 24,9% відповідно;
- аритмія – у 52,9 та 26,0% відповідно;
- ураження клапанів – у 30,3 та 18,2% відповідно.
Отже, найбільш характерною ознакою ураження серця при ХФ є ГМЛШ, яка може бути виявлена за допомогою ЕКГ, ехокардіографії, магнітно-резонансної томографії (МРТ). МРТ серця з пізнім контрастуванням гадолінієм дозволяє виявити фіброз міокарда, що особливо важливо для жінок із ХФ, в яких часто розвивається фіброз у задньобоковій стінці за відсутності ГМЛШ, і МРТ є єдиною можливістю виявити кардіоміопатію Фабрі. Холтерівське моніторування ЕКГ упродовж 48 годин дозволяє виявити скорочення інтервалу PR, АВ-блокаду, зниження варіабельності серцевого ритму, аритмії. Щодо лабораторних показників, при обстеженні пацієнтів із ХФ можуть привернути увагу зниження ШКФ, протеїнурія, підвищення високочутливого тропоніну (Hagege et al., 2019).
У рекомендаціях Європейського товариства кардіологів (ESC, 2014) із діагностики та ведення пацієнтів з гіпертрофічною кардіоміопатією ХФ розглядається як один із найчастіших спадкових метаболічних розладів, що спричиняють розвиток гіпертрофічної кардіоміопатії (ГКМП). Вважається, що на ХФ припадає 0,5-1,0% випадків ГКМП серед осіб старше 35-40 років. Водночас метааналіз 20 досліджень зі скринінгом ХФ свідчить, що ХФ може бути причиною ГКМП у більшій кількості випадків – 0,9-3,9% чоловіків та 1,1-11,8% жінок (Mehta et al., 2006). Поширеність ХФ залежить від віку дебюту ГКМП: у чоловіків вона становить 6,3% при ГКМП, діагностованій у віці >40 років, та 1,4% при ГКМП, виявленій у віці <40 років; у жінок із пізно встановленим діагнозом ГКМП (середній вік – 50 років) поширеність ХФ досягає 12% (Wilcox et al., 2008).
Відповідно до рекомендацій, у дорослих ГКМП діагностується при збільшенні товщини стінки ЛШ ≥15 мм одного або більше сегментів міокарда ЛШ. Але також зазначається, що генетичні захворювання (до яких відноситься і ХФ) можуть проявлятися меншою виразністю потовщення стінки (13-14 мм), і в цих випадках діагноз ГКМП потребує вивчення додаткового матеріалу, зокрема сімейного анамнезу, екстракардіальних симптомів і особливостей, порушень на ЕКГ, проведення лабораторних досліджень і мультимодальної візуалізації серця.
Незважаючи на вказані вище специфічні ознаки та частий дебют ХФ у дитячому віці, діагноз встановлюється пізно (у віці 20-30 років і старше). У значної частини пацієнтів захворювання лишається недіагностованим через брак обізнаності та низьку настороженість лікарів, насамперед кардіологів, нефрологів та неврологів. За даними реєстру Фабрі у США, до встановлення діагнозу пацієнти відвідують дев’ятьох фахівців, час від появи перших симптомів до його визначення може становити до 15 років (Patel, 2011).
Згідно з даними C.M. Eng et al. (2006), пацієнтам із ХФ виставляються різноманітні хибні діагнози: ревматоїдний артрит, ревматична лихоманка, фіброміалгія, синдром хронічної втоми, розсіяний склероз, хронічна демієлінізуюча поліневропатія, невроз, симуляція, синдром Рейно, кардит, автоімунна хвороба, захворювання сполучної тканини, системний червоний вовчак, гострий апендицит, петехії, вівчакові ангіокератоми, васкуліт, невралгія, зловживання лікарськими засобами.
Сергій Андрійович рекомендує запідозрити ХФ за наявності хоча б однієї ознаки з перелічених (Mauer et al., 2018):
- Періодичні епізоди пекучого болю в кінцівках.
- Шкірні прояви ураження судин (ангіокератоми).
- Зниження потовиділення (гіпо- та ангідроз).
- Характерне помутніння рогівки.
- Постпрандіальний біль у животі, нудота та/або діарея невідомого ґенезу в молодому віці.
- ГМЛШ (≥13 мм) або гіпертрофічна кардіоміопатія невідомої етіології, особливо у молодих людей.
- Аритмії невідомої етіології, особливо в молодих пацієнтів.
- Інсульт невідомої етіології в будь-якому віці.
- Хронічна хвороба нирок та/або протеїнурія невідомого ґенезу.
- Випадково виявлені множинні кисти ниркового синуса.
У разі підозри на ХФ для підтвердження діагнозу необхідно визначити ферментну активність α-GLA у плазмі, лейкоцитах, сухих плямах крові (DBS) та провести молекулярно-генетичний аналіз.
Вимірювання активності α-GLA та її відповідне зниження або повна відсутність є золотим стандартом діагностики ХФ у чоловіків. У жінок нормальні показники рівня активності α-GLA не виключають наявності ХФ через особливості інактивації Х-хромосоми. Для підтвердження діагнозу необхідне проведення молекулярно-генетичного аналізу (секвенування ДНК, виявлення мутації гену GLA). Остаточне встановлення діагнозу в одного пацієнта дозволяє виявити у середньому п’ять хворих на ХФ у цій же родині. Істотну роль відіграють збирання сімейного анамнезу та складання генеалогічного древа (Laney et al., 2008).
Простим та доступним методом діагностики є скринінг за методом DBS. Карти для забору біоматеріалу та лабораторну діагностику надає ТОВ «Санофі-Авентіс Україна». Для пацієнтів обстеження абсолютно безкоштовне. Ферментна діагностика проводиться в Метаболічній лабораторії ARCHIMED Life Science GmbH Laboratories (Австрія) або в лабораторії Центру орфанних захворювань НДСЛ «ОХМАТДИТ» (м. Київ). Підставою для остаточного встановлення діагнозу і призначення лікування є виключно генетичне обстеження, яке виконують (після виявлення зниження активності ферменту) в лабораторії ARCHIMED Life Science GmbH Laboratories (Австрія).
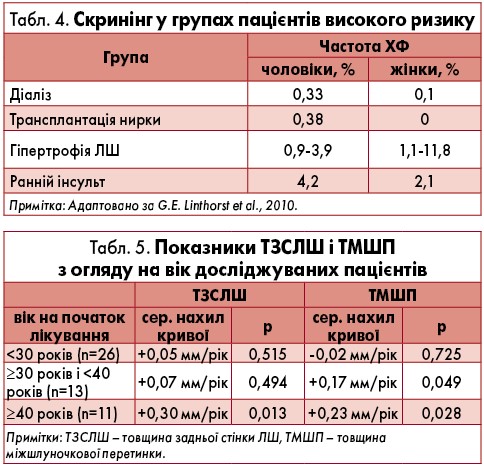
Лектор наголосив на тому, що своєчасний діагноз може врятувати життя хворого. Значна кількість пацієнтів високого ризику з однією із провідних клінічних ознак ХФ може бути ідентифікована за допомогою скринінгу з використанням ферментодіагностики (табл. 4).
 Покращення прогнозу в пацієнтів із ХФ стало можливим з 2001 р., коли для специфічного патогенетичного лікування почали успішно застосовувати замісну терапію рекомбінантними препаратами α-GLА. Оптимальне лікування ХФ включає в себе як патогенетичну терапію, так і симптоматичні методи й лікарські засоби (знеболювальні препарати, гіпотензивна терапія, діаліз, трансплантація нирок, антидепресанти, лікування серцево-судинних ускладнень відповідно до існуючих стандартів). Раннє встановлення діагнозу та початок патогенетичної терапії принципові, оскільки накопичення GL-3 починається з періоду внутрішньоутробного розвитку, а множинне ураження тканин та органів має прогресуючий характер та може бути незворотним (Eng et al., 2008).
Покращення прогнозу в пацієнтів із ХФ стало можливим з 2001 р., коли для специфічного патогенетичного лікування почали успішно застосовувати замісну терапію рекомбінантними препаратами α-GLА. Оптимальне лікування ХФ включає в себе як патогенетичну терапію, так і симптоматичні методи й лікарські засоби (знеболювальні препарати, гіпотензивна терапія, діаліз, трансплантація нирок, антидепресанти, лікування серцево-судинних ускладнень відповідно до існуючих стандартів). Раннє встановлення діагнозу та початок патогенетичної терапії принципові, оскільки накопичення GL-3 починається з періоду внутрішньоутробного розвитку, а множинне ураження тканин та органів має прогресуючий характер та може бути незворотним (Eng et al., 2008).
Рекомбінантна людська агалcидаза бета (препарат Фабразим®*, Sanofi Genzyme) продукується лінією клітин яєчника китайського хом’яка (амінокислотна та нуклеотидна послідовність ідентична натуральній формі α- GLA) та вводиться з розрахунку 1 мг/кг в/в кожні два тижні з максимальною швидкістю інфузії 0,25 мг/хв. Терапевтичної мети вдається досягти завдяки:
- підвищенню кліренсу GL‑3 із ключових місць накопичення;
- стабілізації функції нирок;
- регресії ГМЛШ, покращенню функції міокарда;
- зменшенню больового синдрому;
- поліпшенню якості життя.
Лікування препаратом Фабразим® у дозі 1 мг/кг/2 тижні протягом періоду з медіаною тривалості 10 років зумовлювало сприятливіші наслідки з боку серця у тих пацієнтів, які почали його у більш ранньому віці (Germain et al., 2015). Під час цього випробування вивчали віддалені наслідки терапії у 52 з 58 осіб із класичною формою ХФ. Були використані дані, отримані впродовж дослідження агалсидази бета III фази, 54-місячного розширеного клінічного випробування та реєстру ХФ.
Пацієнтів було розподілено на дві групи залежно від ступеня ураження нирок на вихідному рівні. Медіана періоду спостереження становила 10 років для загальної групи учасників дослідження (n=52). Загалом протягом 10-річного (медіана) періоду лікування не спостерігалося статистично значущого зростання середніх показників товщини задньої стінки ЛШ і товщини міжшлуночкової перетинки (табл. 5). Проте хворі молодшого віку мали сприятливішу відповідь на терапію з боку серця (Germain et al., 2015).
Результати досліджень продемонстрували, що пацієнти, які почали лікування у більш ранньому віці або до розвитку фіброзу, який можливо виявити, отримували найкращий ефект від лікування препаратом Фабразим® у дозуванні 1 мг/кг/2 тижні.
До ризиків застосування агалcидази бета можна віднести формування антитіл протягом трьох місяців після почату терапії, у 50% пацієнтів з антитілами розвиваються асоційовані з інфузією реакції, зокрема озноб та гарячка. Однак згодом титр антитіл перестає наростати, знижується чи зникає повністю. У невеликої частки хворих розвиваються реакції гіперчутливості. До того ж препарат не проникає через гематоенцефалічний бар’єр (Weidemann et al., 2009).
Таким чином, ХФ – життєзагрозливий стан із прогресуючим мультиорганним ураженням, що зустрічається як у чоловіків, так і жінок. Ранні діагностика й початок патогенетичної терапії є критичними для досягнення лікувальних цілей, уповільнення прогресування хвороби, зниження ризику явищ, що становлять загрозу для життя. На сьогодні у нашій країні ХФ безкоштовно діагностується за допомогою вимірювання ферментативної активності й генотипування та може ефективно лікуватися препаратом ФЗТ, що став доступним завдяки гуманітарній програмі Sanofi Genzyme та державній програмі. Активне проведення скринінгу й виявлення пацієнтів із ХФ є важливою, перспективною та ефективною стратегією, якої мають дотримуватись лікарі практично всіх спеціальностей.
Ферментозамісна терапія хвороби Фабрі: нові дані щодо ефективності різних дозувань
Мета дослідження
Оцінити клінічну стабільність та безпеку після переведення пацієнтів з агалсидази бета в дозуванні 11 мг/кг/2 тижні на агалсидазу альфа по 0,2 мг/кг/2 тижні, а також результати після зворотного переходу на агалсидазу бета в дозуванні 1 мг/кг/2 тижні.
Дизайн дослідження
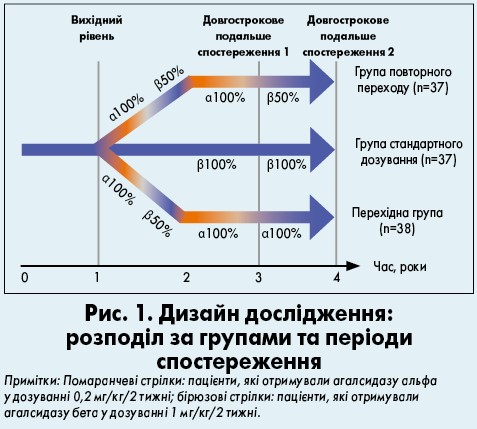
У процесі проспективного обсерваційного дослідження 112 пацієнтів (43 з яких були жіночої статі), що були стабільними на агалсидазі бета 1 мг/кг/2 тижні протягом щонайменше 12 місяців, у нерандомізованому порядку розподілили на три групи; спостереження проводили протягом 53 (діапазон – 38‑57) місяців (рис. 1):
- група стандартного дозування: 37 хворих продовжували отримувати агалсидазу бета у дозуванні 1 мг/кг/2 тижні;
- перехідна група: 38 пацієнтів отримували знижену дозу агалсидази бета і згодом переходили на агалсидазу альфа в дозуванні 0,2 мг/кг/2 тижні або одразу ж – на агалсидазу альфа по 0,2 мг/кг/2 тижні і лишалися на агалсидазі альфа 0,2 мг/кг/2 тижні;
- група повторного переходу: 37 хворих знову переводили на агалсидазу бета у дозуванні 1 мг/кг/2 тижні після того, як вони отримували агалсидазу альфа по 0,2 мг/кг/2 тижні протягом щонайменше 12 місяців.
Середня тривалість терапії агалсидазою бета у дозуванні 1 мг/кг/2 тижні до включення пацієнта в дослідження становила в середньому 31 (12‑60) місяць. Дані хворого за попередній період використовували як контрольні, що давало можливість проаналізувати індивідуальні наслідки зміни лікування.
Вихідні характеристики
Пацієнти, які продовжували отримувати агалсидазу бета у дозуванні 1 мг/кг/2 тижні протягом усього дослідження, мали тяжчі ураження, ніж у перехідній групі та групі повторного переходу:
достовірно більше хворих чоловічої статі, у яких була нижча активність α-галактозидази A (GLA) порівняно з іншими групами (обидва р<0,05);
частіші симптоми, пов’язані з хворобою Фабрі (такі як ангіокератоми, діарея, гіпогідроз та напади болю, асоційованого з хворобою Фабрі), порівняно з пацієнтами перехідної групи (р<0,05);
більша кількість балів за шкалою Майнца для оцінки ступеня тяжкості захворювання (MSSI) порівняно з пацієнтами групи повторного переходу (р<0,05).
Ключові результати
Функція нирок
У пацієнтів групи повторного переходу на агалсидазу бета в дозуванні 1 мг/кг/ 2 тижні спостерігалося зменшення значень рШКФ (р<0,05). Щорічне зниження показників рШКФ від періоду подальшого спостереження 1 (ППС‑1) до періоду подальшого спостереження 2 (ППС‑2) становило -4,6 мл/хв/1,73 м2 у перехідній групі порівняно з -2,2 мл/хв/1,73 м2 у групі повторного переходу. У хворих, які отримували стандартне дозування, рівень рШКФ лишався стабільним протягом усього періоду подальшого спостереження (рис. 2).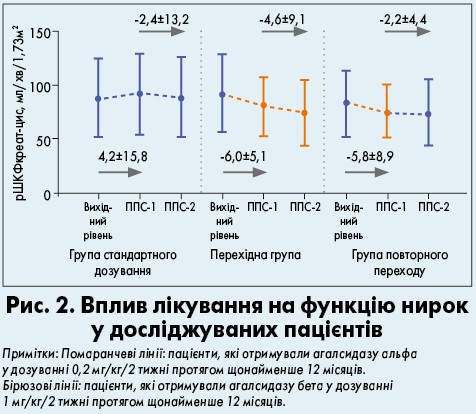
Гастроінтестинальні симптоми
Повторний перехід на агалсидазу бета у дозуванні 1 мг/кг/2 тижні приводив до значного зниження частоти випадків діареї (р<0,05). У пацієнтів перехідної групи частіше повідомлялося про діарею під час першого довгострокового періоду подальшого спостереження (р<0,05). У хворих, які отримували регулярні дози, симптоми з боку шлунково-кишкового тракту залишалися стабільними протягом усього періоду подальшого спостереження.
Lyso-GL‑3
У групі повторного переходу на агалсидазу бета 1 мг/кг/2 тижні спостерігалося значне зниження рівнів глоботріаозилсфінгозину (Lyso-GL‑3); р<0,05 (рис. 3). Рівні Lyso-GL‑3 лишалися стабільними між ППС‑1 і ППС‑2 у групі стандартного дозування та перехідній. У пацієнтів перехідної групи відзначалися найвищі значення Lyso-GL‑3 під час ППС‑1 (р<0,05, порівняно з групами стандартного дозування і повторного переходу).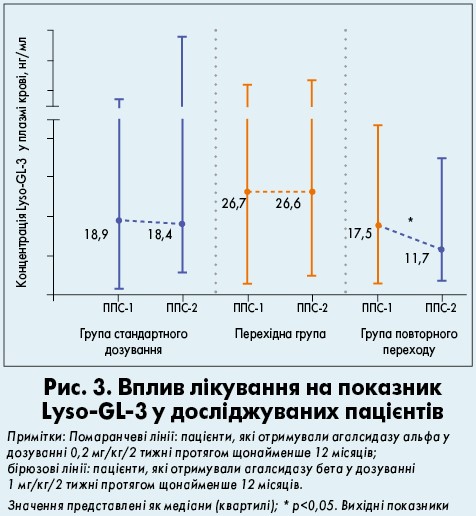
Безпека
Повторний перехід на агалсидазу бета у дозуванні 1 мг/кг/2 тижні добре переносився і не призводив до клінічно значущих інфузійних побічних реакцій. У трьох (8%) пацієнтів групи повторного переходу спостерігалися незначні інфузійні реакції (несуттєве підвищення температури тіла і надмірна стомлюваність).
Висновки
Повторний перехід на агалсидазу бета у дозуванні 1 мг/кг/2 тижні мав позитивний вплив на функцію нирок, симптоми з боку шлунково-кишкового тракту та рівні Lyso-GL‑3. У пацієнтів, які були повторно переведені на агалсидазу бета в дозуванні 1 мг/кг/2 тижні, спостерігалися:
- уповільнення погіршення функції нирок;
- статистично значуще зниження частоти випадків діареї;
- статистично значуще зменшення рівнів lyso-GL‑3 у плазмі крові.
Хворі перехідної групи мали постійне зниження рівнів рШКФ і частіше повідомляли про діарею. У пацієнтів, які отримували агалсидазу бета у дозуванні 1 мг/кг/2 тижні, частота випадків діареї, бали за MSSI та показники функції нирок залишалися стабільними.
Адаптовано за J. Krаmer et al., 2017; doi: 10.1093/ndt/gfx319.
*Лікарський засіб Фабразим®, порошок для приготування концентрату 5 мг/мл для розчину для інфузій, зареєстрований в Україні. Р.П. № UA/10306/01/01. Наказ МОЗ України № 2352 від 28.11.2019.
Підготувала Маргарита Марчук
Тематичний номер «Кардіологія, Ревматологія, Кардіохірургія» № 2 (69) 2020 р.
СТАТТІ ЗА ТЕМОЮ Кардіологія
Дисліпідемія та атеросклеротичні серцево-судинні захворювання (АСССЗ) є провідною причиною передчасної смерті в усьому світі (Bianconi V. et al., 2021). Гіперхолестеринемія – третій за поширеністю (після артеріальної гіпертензії та дієтологічних порушень) фактор кардіоваскулярного ризику в світі (Roth G.A. et al., 2020), а в низці європейських країн і, зокрема, в Польщі вона посідає перше місце. Актуальні дані свідчать, що 70% дорослого населення Польщі страждають на гіперхолестеринемію (Banach M. et al., 2023). Загалом дані Польщі як сусідньої східноєвропейської країни можна екстраполювати і на Україну....
Сечова кислота (СК) – кінцевий продукт метаболізму пуринів, який здебільшого синтезується в печінці та виводиться нирками і кишечником [1, 2]. Гіперурикемія – підвищений рівень СК у сироватці крові – є метаболічною основою подагри, одного з найпоширеніших запальних артритів. У середні віки подагра вважалася хворобою надмірності аристократії, нині її поширеність зростає у всьому світі через зміни в харчуванні, переважання в дієті оброблених продуктів, фруктози та збільшення поширеності ожиріння [3]....
Глюкокортикоїди (ГК), які використовуються з початку 1950-х рр., стали невід’ємною частиною лікування ревматоїдного артриту (РА) [1]. Ключовою перевагою ГК є швидке настання ефекту, особливо порівняно з класичними синтетичними хворобомодифікувальними антиревматичними препаратами (ХМАРП) на кшталт метотрексату. Відповідно, ГК мають привабливий профіль для лікування спалахів хвороби або для застосування в ролі засобів для бридж-терапії на ранніх стадіях РА в очікуванні ефекту класичних синтетичних ХМАРП. Ця стратегія широко використовується після публікації результатів дослідження COBRA в 1997 р. [2]. ...
Гостре розтягнення зв’язок гомілковостопного суглоба (ГРЗГС) є поширеним, інвалідизувальним, клінічно значущим захворюванням, щодо якого по медичну допомогу щороку звертається >1 млн пацієнтів [1]; асоціюється зі значним соціально-економічним тягарем [2]. Майже 16-40% випадків ГРЗГС – спортивна травма, а час одужання є дуже важливим для спортсменів, професіоналів та осіб, котрі готуються до великих змагань. Найпоширенішими (85%) є латеральні (бічні) розтягнення зв’язок гомілковостопного суглоба; особливо часто ушкоджується передня таранно-гомілкова зв’язка – зазвичай унаслідок високошвидкісної інверсії та внутрішньої ротаційної травми [3]; ≈40% розтягнень гомілковостопного суглоба мають ризик хронізації. Характерними ознаками ГРЗГС є біль під час навантаження, нестабільність гомілковостопного суглоба та проблеми з рухливістю [4-6]....