



Клінічні та морфологічні особливості первинних CD30+ лімфопроліферативних захворювань шкіри
 Випадки з клінічної практики дерматопатолога
Випадки з клінічної практики дерматопатолога
Т-клітинні лімфоми шкіри (ТКЛШ) – це широка група захворювань, яка включає різні нозології, що характеризуються специфічними клінічними проявами, морфологічними змінами та прогнозом.
Лімфоматоїдний папульоз (ЛП) і первинна анапластична великоклітинна лімфома шкіри (ПШ-АВЛ) є складовими одного патологічного процесу та мають подібні морфологічні та імуногістохімічні характеристики, проте різну клінічну картину [1].
Наявність пограничних станів, коли у пацієнта спостерігаються прояви обох захворювань, підтверджує цю теорію. Тому в новій класифікації лімфом шкіри [2] ЛП і ПШ-АВЛ включені у групу первинних CD30+ лімфопроліферативних захворювань, що характеризуються експресією на поверхні клітин антигена CD30 та, незважаючи на агресивні морфологічні ознаки, мають сприятливий прогноз. CD30 також експресується при низці інших ТКЛШ, тому діагностика таких станів потребує повного імунофенотипування клітин інфільтрату та ретельної клініко-морфологічної кореляції.
Клінічний випадок 1
Пацієнтка, 32 роки, звернулася в лікарню зі скаргами на пухлину з виразкою (рис. 1А), що виникла кілька місяців тому. Пацієнтка також відзначала появу папул та вузлів (рис. 1Б), що іноді супроводжувалися виразкуванням, а згодом самостійно загоювалися з утворенням нормотрофічних рубців. Загальний стан пацієнтки на момент огляду задовільний. Хворій проведено біопсію, за результатами якої виявлено такі зміни (рис. 1В): у дермі спостерігався набряк та дифузний лімфоцитарний інфільтрат із малих та середніх лімфоцитів зі значною домішкою великих клітин, які мали гіперхромні великі ядра, визначалась велика кількість фігур мітозів. Дані імуногістохімічного дослідження: клітини пухлини CD4, CD30-позитивні, частина клітин – CD3-позитивні та ALK-негативні (останні не представлені на рисунку) (рис. 1Г-Е).
Після того, як на підставі клініко-лабораторних даних було виключено системну АВЛ, за морфологічними змінами та імунофенотипом клітин пухлини діагностовано ПШ-АВЛ, однак, враховуючи клінічний перебіг та наявність висипу, що самостійно загоювався та з’являвся знову, цей випадок захворювання можна віднести до пограничних станів, коли у пацієнта наявні прояви як ПШ-АВЛ, так і ЛП.


Клінічний випадок 2
Пацієнтка, 52 роки, мала скарги на папуло-некротичний висип на тулубі, який суб’єктивно не турбував (рис. 2А-Б), відзначала появу нових елементів висипу з одночасним регресуванням старих. Клінічний діагноз – pityriasis lichenoides et varioloformis acuta. Морфологічні ознаки захворювання – зливний пара- та гіперкератоз, визначалися лейкоцити у паракератотичних масах. У базальних відділах епідермісу спостерігався інтерфейс-дерматит із наявністю цитоїдних тілець та сателітних некрозів кератиноцитів (рис. 2В). При імуногістохімічному дослідженні серед клітин інфільтрату виявлено CD30+ клітини (рис. 2Г). На підставі наявності характерної клінічної картини та морфологічних змін встановлено діагноз лімфоматоїдного папульозу (тип В).
Клінічний випадок 3
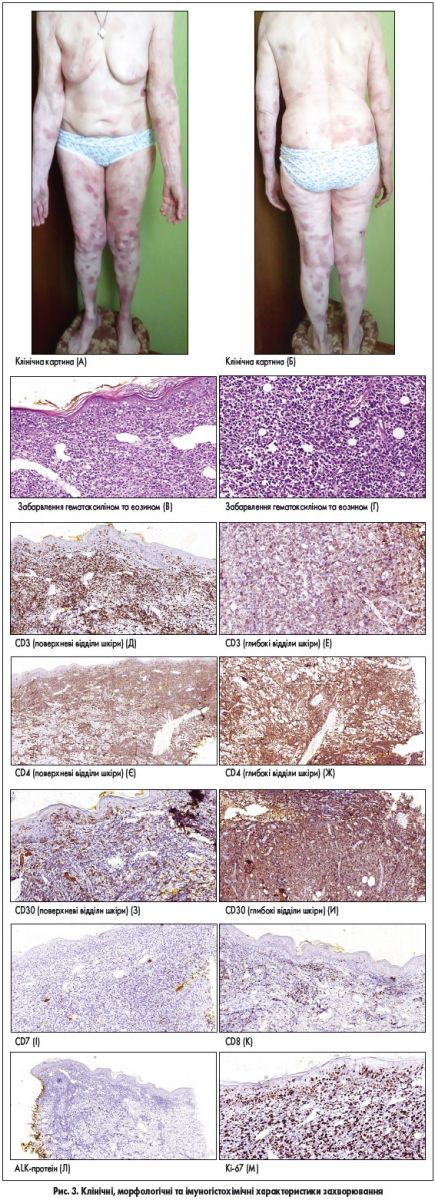
Пацієнтка, 72 роки, звернулася в лікарню зі скаргами на плями на шкірі, які спостерігалися протягом тривалого періоду та суб’єктивно не турбували до останнього часу, проте кілька місяців тому на місці плям з’явилися спочатку бляшки, а згодом – виразки (рис. 3А-Б). При біопсії в дермі спостерігався щільний лімфоцитарний інфільтрат переважно із малих та середніх лімфоцитів зі значною домішкою великих гіперхромних клітин, що розміщувались як у поверхневих, так і у глибоких відділах дерми з поширенням у підшкірну жирову клітковину (рис. 3В-Г). Визначено також виражений епідермотропізм клітин інфільтрату з формуванням в епідермісі дрібних скупчень (мікроабсцесів Потріє) (рис. 3В). Імунофенотип клітин інфільтрату – CD3+, CD4+ та CD30+, CD7-, CD8-, ALK-. Приблизно 80% клітин пухлини Ki‑67-позитивні (маркер проліферації; рис. 3Д-М). На підставі наявної морфологічної картини та визначення імунофенотипу клітин пухлини, а також клінічних даних діагностована великоклітинна трансформація грибоподібного мікозу.
 Обговорення
Обговорення
ТКЛШ – широка група захворювань, яка включає різні нозології, що характеризуються специфічними клінічніми проявами, морфологічними змінами та прогнозом [2] (табл. 1).
Найбільш поширеними у групі ТКЛШ є грибоподібний мікоз та синдром Сезарі, які становлять приблизно 65% усіх випадків. CD30+ лімфопроліферативні захворювання, такі як ЛП та ПШ-АВЛ, – близько 25%. Частота інших лімфом, що зустрічаються значно рідше, становить лише 10% випадків (табл. 2).
Залежно від клінічного перебігу ТКЛШ поділяють на агресивні та індолентні (табл. 3).
Незважаючи на те що грибоподібний мікоз належить до групи індолентних лімфом, при великоклітинній трансформації прогноз цього захворювання значно погіршується [4].
Первинні CD30+ лімфопроліферативні захворювання шкіри
Первинні CD30+ лімфопроліферативні захворювання шкіри складають другу за поширеністю групу нозологій серед ТКЛШ [2] (табл. 2). До них належать ПШ-АВЛ, ЛП та пограничні стани.
 ЛП та ПШ-АВЛ можуть мати спільні клінічні, морфологічні та імуногістохімічні ознаки. На даному етапі вивчення вважається, що ці захворювання складають спектр одного патологічного процесу [5].
ЛП та ПШ-АВЛ можуть мати спільні клінічні, морфологічні та імуногістохімічні ознаки. На даному етапі вивчення вважається, що ці захворювання складають спектр одного патологічного процесу [5].
Для встановлення морфологічного діагнозу пацієнтам із підозрою на первинні CD30+ лімфопроліферативні захворювання шкіри необхідно проводити біопсію з подальшим імуногістохімічним дослідженням та за необхідності молекулярно-генетичне дослідження для встановлення клональності клітин інфільтрату.
Лімфоматоїдний папульоз
Вперше це захворювання описав W.L. Macaulay в 1968 році [6]. У структурі захворюваності ТКЛШ частка ЛП становить 15%. ТКЛШ уражує осіб усіх вікових груп, середній вік пацієнтів становить 35-45 років. У чоловіків захворювання реєструють частіше, ніж у жінок (1,5:1). ЛП – хронічне доброякісне самолімітуюче захворювання, що характеризується папульозним, папуло-вузловим чи папуло-некротичним висипом та має рецидивуючий характер. Висип локалізований, як правило, дисеміновано на кінцівках і тулубі пацієнта, однак може мати і згрупований характер, обмежуючись однією локалізацією. Колір елементів висипу варіює від червоного до фіолетового, розмір – від декількох міліметрів до 2 см. Оскільки висип має рецидивуючий характер, із часом у пацієнта починає спостерігатися поліморфізм елементів. У приблизно 50% випадків захворювання висип суб’єктивно не турбує, але іноді наявні свербіння та відчуття печіння. Зазвичай висип зникає протягом 1-4 міс, але може спостерігатися протягом тривалого часу – декілька місяців та навіть років, найдовший зафіксований період його наявності становив близько 40 років [7].
Патогенез ЛП поки що не з’ясований. Декілька авторів висунули припущення про вірусну природу цього захворювання, проте за результатами низки досліджень не встановлено роль Т-лімфотропного вірусу людини типу 1 (HTLV‑1), вірусу Епштейна – Барр (EBV) та інших вірусів герпесвірусної групи у патогенезі ЛП. Механізм спонтанної регресії висипу, що спостерігається у частини пацієнтів, до цього часу не встановлений. Найбільш ймовірно, причиною цього феномену є взаємодія CD30 з лігандом CD30, що призводить до апоптозу пухлинних Т-клітин [8].
Виділяють 5 основних морфологічних типів ЛП – типи A-E. Нещодавно були описані: 6-й – тип F та рідкісні типи, такі як γ/δ-тип та ЛП із реаранжируванням генів 6р25 [10].
При імуногістохімічному дослідженні у більшості випадків визначаються клітини інфільтрату CD45RO+ та CD4+. Усі форми захворювання характеризуються наявністю CD30+ клітин в інфільтраті, за винятком типу В, який потребує ретельної диференційної діагностики з грибоподібним мікозом. У цьому випадку важливим є проведення клініко-морфологічної кореляції. Типи D, E ЛП у дітей можуть бути CD4-, CD8+. Важливо диференціювати ЛП, який є CD45RO-позитивним, з епідермотропною CD8+ ТКЛШ, яка зазвичай CD45RО- [2].
Окрім цього, при проведенні диференційної діагностики слід враховувати можливість наявності станів, перелічених у таблиці 4.
Слід зазначити, що характер клінічного перебігу не залежить від морфологічного підтипу захворювання. Його визначення має важливе значення для проведення диференційної діагностики та оцінки прогнозу, оскільки підтипи В та С асоційовані з розвитком вторинних гематологічних пухлин [11].
Первинна анапластична великоклітинна лімфома шкіри
ПШ-АВЛ характеризується наявністю солітарної пухлини розміром >2 см у діаметрі, яка швидко росте та супроводжується виразкуванням. Іноді трапляються багатовогнищеві форми захворювання. Незважаючи на агресивний клінічний перебіг ПШ-АВЛ, пацієнти мають добре самопочуття, у них не спостерігаються симптоми системного ураження (температура, слабкість, підвищена пітливість, зменшення маси тіла та ін.). Наявність цих симптомів має велике значення, тому що вкрай важливим у діагностиці ПШ-АВЛ є виключення можливого вторинного ураження шкіри внаслідок системної АВЛ. Близько 20% пацієнтів із системною АВЛ мають вогнища вторинного ураження шкіри [12].
При морфологічному дослідженні захворювання характеризується щільним лімфоцитарним інфільтратом з анапластичною морфологією: клітини мають рясну еозинофільну цитоплазму та великі, плеоморфні, гіперхромні ядра, деякі мають форму підкови та велике еозинофільне ядерце. Для встановлення діагнозу ПШ-АВЛ щонайменше 75% клітин пухлини мають бути CD30-позитивними. При шкірних формах захворювання кіназа анапластичних лімфом (ALK) зазвичай негативна, однак це не має бути вирішальною ознакою при диференціюванні зі системною АВЛ, оскільки близько 50% цих пухлин є ALK- [13]. Шкірний лімфоцитарний антиген (CLA) при ПШ-АВЛ, як правило, позитивний, натомість епітеліальний мембранний антиген (EMA) – негативний [14].
Проводити диференційну діагностику необхідно зі системною АВЛ, ЛП, великоклітинною трансформацією грибоподібного мікозу та іншими лімфомами, клітини яких можуть експресувати на своїй поверхні CD30 антиген (у тому числі з вторинним ураженням шкіри при лімфомі Ходжкіна), а також із реактивними запальними реакціями.
Пограничні стани
До пограничних станів належать випадки захворювання, якщо диференціювати ЛП та ПШ-АВЛ неможливо. За результатами ретельного і тривалого спостереження пацієнта можна більш точно визначити, яке з цих захворювань має місце.
Прогноз захворювання
Первинні CD30+ лімфопроліферативні захворювання шкіри мають сприятливий прогноз. Десятирічна виживаність становить 85-100% [14]. При веденні таких пацієнтів слід враховувати, що ЛП може бути асоційований з розвитком вторинних гематологічних пухлин. Найчастіше спостерігаються грибоподібний мікоз та АВЛ [11].
Залежно від діагностованої форми захворювання клінічна тактика при лікуванні ТКЛШ та прогноз значно відрізняються порівняно з нодальною формою. Наприклад, на відміну від останньої, ПШ-АВЛ не потребує агресивного лікування, тому встановлення точного діагнозу є запорукою вибору правильної тактики ведення пацієнта.
Загальні відомості про антиген CD30
Антиген CD30 (кластер диференціювання 30) – трансмембранний глікопротеїн 1 типу, який належить до суперсімейства рецепторів фактора некрозу пухлини 8 (TNFRSF8). Вперше цей рецептор був описаний у клітинах лімфоми Ходжкіна, згодом аналогічний антиген виявлено у низці неходжкінських лімфом, у тому числі ПШ-АВЛ.
CD30 – мембранний цитокіновий рецептор, що з’являється на поверхні мітогенактивованих Т- та деяких В-лімфоцитів. Експресія CD30 потребує активації сигнального шляху CD28 чи рецептора інтерлейкіну-4 [15, 16].
CD30 взаємодіє з лігандом CD30 (CD30L, CD153, TNFSF8) – мембранним глікопротеїном 2 типу, що також належить до суперсімейства TNF та експресується на активованих Т-лімфоцитах, переважно на поверхні CD4+ клітин – Т-хелперах 1 та 2 типу, а також на поверхні В-лімфоцитів та деяких інших імунокомпетентних клітин, у тому числі антигенпрезентуючих клітин [17]. Активація сигнального шляху CD30 призводить до активації ядерного фактора NF-kB як шляхом активації TNF-рецептор-асоційованого фактора 2 (TRAF), так і через незалежний від цього фактора шлях, що може пригнічувати активність ефекторних клітин, стимулювати апоптоз або, навпаки, призводити до виживання клітин залежно від типу залучених до активації клітин та активованих сигнальних шляхів [18, 19].
 Експресію CD30 на поверхні лімфоцитів можуть спричиняти ціла низка факторів, у тому числі реактивні стани, запальні та інфекційні захворювання, пухлинні процеси, тому для встановлення діагнозу необхідними є ретельна клініко-морфологічна кореляція, проведення морфологічного та імуногістохімічного дослідження біоптатів шкіри, а в деяких випадках – молекулярно-генетичного дослідження з визначенням клональності Т-клітин інфільтрату [20] (табл. 4).
Експресію CD30 на поверхні лімфоцитів можуть спричиняти ціла низка факторів, у тому числі реактивні стани, запальні та інфекційні захворювання, пухлинні процеси, тому для встановлення діагнозу необхідними є ретельна клініко-морфологічна кореляція, проведення морфологічного та імуногістохімічного дослідження біоптатів шкіри, а в деяких випадках – молекулярно-генетичного дослідження з визначенням клональності Т-клітин інфільтрату [20] (табл. 4).
Висновки
ТКЛШ – широка група захворювань, яка включає різні нозології, що характеризуються специфічними клінічніми проявами, морфологічними змінами та прогнозом. ЛП та ПШ-АВЛ складають спектр одного патологічного процесу та мають подібні морфологічні та імуногістохімічні характеристики, проте різну клінічну картину.
CD30 – важливий маркер для діагностики та класифікації ЛШ, однак інтерпретацію позитивної реакції слід здійснювати у комплексі з точними імунофенотипічними характеристиками пухлини та вичерпними клінічними даними.
Незважаючи на те що більшість ТКЛШ мають індолентний клінічний перебіг, своєчасне встановлення точного діагнозу має бути правилом, а не винятком. Без повних клінічних даних проведення диференційної діагностики може бути ускладненим або навіть неможливим, тому клініко-морфологічна кореляція є критично важливою у веденні пацієнтів із підозрою на ТКЛШ.
Протягом останніх кількох років спостерігається значний прогрес із точки зору розуміння клінічного перебігу, патологічних характеристик, біологічного потенціалу та етіологічних чинників групи захворювань, до якої належить ТКЛШ. Незважаючи на це, досі є багато нез’ясованих питань, зокрема, активно дискутуються класифікація, етіологічні фактори та прогностичні ознаки цих захворювань.
Література
- Ralfkiaer E., Willemze R., Paulli M., Kadin M.E. Primary cutaneous CD30-positive T-cell lymphoproliferative disorders. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 2008; 300-301.
- WHO-EORTC classification for cutaneous lymphomas // Willemze R., Jaffe E.S., Burg G., Cerroni L., Berti E., Swerdlow S.H. et al. Blood. 2005 (15 May); 105(10): 3768-3785.
- Facchetti F., Petrella T., Pileri S.A. Вlastic plasmacytoid dendritic cell neoplasm. [авт. книги] Campo E., Harris N.L., Jaffe E.S., Pileri S.A., Stein H., Thiele J., Swerdlow S.H. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th. 2017: 174-178.
- Pulitzer M., Myskowski P.L., Horwitz S.M., Querfeld C., Connolly B., Li J. and Murali R. Mycosis fungoides with large cell transformation. Pathology. 2014 December: 46(7): 610-616.
- Primary cutaneous CD30+ lymphoproliferative disorders: new insights into biology and therapy // Querfeld C., Kuzel T.M., Guitart J., Rosen S.T. Oncology. 2007 (1 May); 21(6): 689-96; discussion: 699-700.
- Macaulay W.L. Lymphomatoid papulosis. A continuing self-healing eruption, clinically benign-histologically malignant. Arch Dermatol, 1968 Jan; 97(1): 23-30.
- Rein W. Cutaneous T-Cell Lymphoma. [авт. книги] Jorizzo J., Schaffer J.L., Bolognia J.V. Dermatology, 2012: 2029-2032.
- Mori M., Manuelli C., Pimpinelli N., Mavilia C., Maggi E., Santucci M., Bianchi B., Cappugi P., Giannotti B. and Kadin M.E. CD30-CD30 Ligand Interaction in Primary Cutaneous CD30+T-Cell Lymphomas: A Clue to the Pathophysiology of Clinical Regression. Blood, 1999; 94: 3077-3083.
- Morimura S., Sugaya M., Tamaki Z. et al. Lymphomatoid papulosis showing γδ T-cell phenotype. Acta Derm Venereol. 2011; 91: 712-713.
- Karai L.J., Kadin M.E., Hsi E.D. et al. Chromosomal rearrangements of 6p25.3 define a new subtype of lymphomatoid papulosis. Am J Surg Pathol. 2013; 37: 1173.
- Wieser I., Oh C.W., Talpur R., Duvic M. Lymphomatoid papulosis: Treatment response and associated lymphomas in a study of 180 patients. J Am Acad Dermatol. 2016 Jan; 74(1): 59-67.
- Falini B., Pileri S., Zinzani P.L., Carbone A. et al. ALK+ lymphoma: clinico-pathological findings and outcome. Blood. 1999 (15 Apr); 93(8): 2697-2706.
- Gascoyne R.D., Aoun P., Wu D., Chhanabhai M. et al. Prognostic significance of anaplastic lymphoma kinase (ALK) protein expression in adults with anaplastic large cell lymphoma. Blood. 1999 (1 Jun); 93(11): 3913-3921.
- Primary and secondary cutaneous CD30(+) lymphoproliferative disorders: a report from the Dutch Cutaneous Lymphoma Group on the long-term follow-up data of 219 patients and guidelines for diagnosis and treatment // Bekkenk M.W., Geelen F.A., van Voorst Vader P.C., Heule F., Geerts M.L., van Vloten W.A., Meijer C.J., Willemze R. Blood. 2000 (Jun 15); 95(12): 3653-3661.
- Gilfillan M.C., Noel P.J., Podack E.R., Reiner S.L., Thompson C.B. Expression of the costimulatory receptor CD30 is regulated by both CD28 and cytokines. J Immunol. 1998; 160(5): 2180-2187.
- Smith C.A., Gruss H.J., Davis T., Anderson D., Farrah T., Baker E., Sutherland G.R., Brannan C.I., Copeland N.G., Jenkins N.A. et al. CD30 antigen, a marker for Hodgkin’s lymphoma, is a receptor whose ligand defines an emerging family of cytokines with homology to TNF. Cell. 1993 (2 Jul); 73(7): 1349-1360.
- Bowen M.A., Lee R.K., Miragliotta G., Nam S.Y., Podack E.R. Structure and expression of murine CD30 and its role in cytokine production. J Immunol. 1996; 156: 442-449.
- Song H.Y., Regnier C.H., Kirschning C.J., Goeddel D.V., Rothe M. Tumor necrosis factor (TNF)-mediated kinase cascades: bifurcation of nuclear factor-kappaB and c-jun N-terminal kinase (JNK/SAPK) pathways at TNF receptor-associated factor 2. Proc Natl Acad Sci USA. 1997 (2 Sept); 94(18): 9792-9796.
- Bargou R.C., Emmerich F., Krappmann D., Bommert K., Mapara M.Y., Arnold W. et al; Royer H.D., Grinstein E., Greiner A., Scheidereit C., Dorken B. Constitutive nuclear factor-kappaB-RelA activation is required for proliferation and survival of Hodgkin’s disease tumor cells. J Clin Invest. 1997 (15 Dec); 100(12): 2961-2969.
- LeBoeuf N.R., McDermott S. and Harris N.L. Case 5-2015 – A 69-Year-Old Woman with Recurrent Skin Lesions after Treatment for Lymphoma. N Engl J Med. 2015 (12 February); 372: 650-659.
- Wieser I., Oh C.W., Talpur R., Duvic M. Lymphomatoid papulosis: Treatment response and associated lymphomas in a study of 180 patients. J Am Acad Dermatol. 2016 Jan; 74(1): 59-67.
Всі фото публікуються з дозволу автора.
За підтримки ТОВ «Такеда Україна»: 03110, м. Київ, вул. Солом’янська, 11
тел.: (044) 390 2929, www.takeda.ua
UA/ADC/0718/0024
Тематичний номер «Онкологія» № 3 (54), червень-липень 2018 р.
СТАТТІ ЗА ТЕМОЮ Дерматологія
У лютому відбувся онлайн-майстер-клас «Персоніфікація сучасної медицини. Важливі питання гендерних особливостей перебігу захворювань внутрішніх органів. Вплив психоемоційних та інших складових», організований кафедрою терапії, інфекційних хвороб і дерматовенерології Інституту післядипломної освіти Національного медичного університету ім. О.О. Богомольця (м. Київ), а також ГО «Українська академія функціональної медицини та гастропсихології» (м. Київ). ...
Гостра лімфобластна лейкемія (ГЛЛ) є найпоширенішим онкогематологічним захворюванням у дітей і складає значну частку серед лейкемій у дорослих. Незважаючи на значні успіхи в лікуванні ГЛЛ у дітей, де рівень виліковності сягає 90%, результати терапії у дорослих залишаються незадовільними. У рамках науково-практичної конференції з міжнародною участю «Діагностика та лікування гематологічних захворювань: підведення підсумків 2023 року» (15-16 грудня 2023 року) проведено секцію, присвячену ГЛЛ....
Хронічна лімфоцитарна лейкемія (ХЛЛ) залишається актуальною проблемою сучасної онкогематології. Незважаючи на певні досягнення в терапії, ХЛЛ є невиліковним захворюванням. Стандартна хіміотерапія не забезпечує стійкої відповіді, а трансплантація гемопоетичних стовбурових клітин можлива лише для окремої когорти пацієнтів. Тому пошук нових підходів до терапії ХЛЛ, зокрема таргетної, є нагальним завданням. ...
Гепатоцелюлярна карцинома (ГЦК) – злоякісне новоутворення в печінці, що розвивається з гепатоцитів. Рання діагностика і початок лікування пацієнтів із ГЦК запобігає виникненню тяжких ускладнень і покращує якість життя пацієнтів. Медична допомога пацієнтам із ГЦК потребує міждисциплінарної співпраці та інтегрованого ведення хворих мультидисциплінарною командою фахівців, яка займається або спеціалізується на злоякісних новоутвореннях печінки. Саме цьому сприятимуть положення Стандарту медичної допомоги «Гепатоцелюлярна карцинома»....