


6 січня, 2020
Роль дефицита альфа1-антитрипсина в развитии бронхолегочной патологии
Дефицит альфа1-антитрипсина (А1АТ) – генетически детерминированное заболевание, вызванное недостаточностью А1АТ в сыворотке крови, возникающее вследствие различных мутаций в гене Pi и проявляющееся в виде хронической обструктивной болезни легких (ХОБЛ), эмфиземы легких, идиопатического фиброза, бронхоэктазов, развития рака легкого, поражения печени и сосудов.
Точная распространенность дефицита А1АТ в большинстве популяций не известна, и у многих он остается не диагностированным. Например, в США из 70-100 тыс. пациентов с признаками дефицита А1АТ идентифицированы только 10% [1], а в Европе из 125 тыс. лиц лишь приблизительно 5 тыс. включены в Международный регистр альфа‑1 (The Alpha One International Registry) [2]. В многочисленных исследованиях было показано, что срок установления диагноза составляет от 5,6 до 7,2 года с момента первого обращения к врачу. Это связано с тем, что у некоторых людей с очень низким уровнем этого белка не отмечаются клинические признаки заболевания, а симптомы различны или полностью отсутствуют. Кроме того, на проявление заболевания влияет не только генетическая предрасположенность, но и факторы окружающей среды. Распространенность дефицита А1АТ в мире не превышает 0,5-1% (наибольшая – 1,8% – в странах Скандинавии). Дефицит А1АТ в организме человека является недооцененным заболеванием.
Физиологическая роль А1АТ
А1АТ имеет альтернативное название «ингибитор альфа‑1-протеиназы», или SERPINA1 (SERine Proteinase INhibitor), является членом суперсемейства ингибиторов сериновых протеаз (серпинов), которое состоит из альфа‑1-химотрипсина, ингибитора С1, антитромбина, нейросерпина и др. [3, 4]. А1АТ считается основной антиэластазой нижних дыхательных путей, в основе действия лежит способность ингибировать сериновую протеазу и нейтрофильную эластазу [5, 6]. Кроме антипротеазной активности, А1АТ обладает другими биологическими эффектами (модулирование воспаления), антиоксидантным и антимикробным действием, участвует в локальном иммунном ответе, препятствует апоптозу [7, 8]. Мутантные формы А1АТ играют важную роль в патогенезе заболеваний. Это связано с неправильным сворачиванием белка, аккумулированием этих форм в полимеры, что приводит к уменьшению секреции функциональных мономеров.
История открытия А1АТ
В 1963 г. ученые C.-B. Laurell и S. Eriksson из Лундского университета в Швеции обнаружили, что при электрофорезе белков сыворотки у лиц с панацинарной эмфиземой не было полосы для альфа‑1-глобулина [9, 10].
Через шесть лет, в 1969 г., H. Sharp и соавт. [11] установили связь между дефицитом А1АТ и цирротическими изменениями при поражении печени.
Структура и функциональная роль А1АТ
А1АТ представляет собой гликопротеин с молекулярным весом 52 кДа, который секретируется гепатоцитами и в меньшей степени другими тканями, включая эпителиальные клетки легкого, макрофаги, тубулярные клетки почек, кишечные эпителиальные клетки. А1АТ относится к быстросинтезируемым белкам (<90 мин), а период полувыведения составляет ~4-5 дней. Функция А1АТ заключается в ингибировании химотрипсина, панкреатической эластазы, коллагеназы кожи, ренина, урокиназы, фактора Хагемана и нейтральных протеаз нейтрофилов. Активная форма А1АТ состоит из 394 аминокислот и имеет сложное строение – три определенным образом упакованные β-структуры, девять α-спиралей и ингибиторный метионин реактивного центра (Met358). Взаимодействие А1АТ с протеазой начинается с того, что она подвергает атаке метионина реактивного центра ингибитора антитрипсина. Остаток метионина реактивного центра А1АТ служит «приманкой» для фермента. Образовавшийся комплекс претерпевает конформационные изменения, в результате которых протеаза погружается вглубь молекулы А1АТ и в дальнейшем активируется [12]. Распространяясь с кровотоком, А1АТ попадает в легкие путем диффузии через эндотелий и эпителиальные клетки. На поверхности бронхиального эпителия А1АТ определяется в количестве 10-15% от содержания в плазме. Данный белок присутствует в слезах, дуоденальной жидкости, слюне, выделениях из носа, спинномозговой жидкости, выделениях из легких, материнском молоке. Это острофазный белок, уровень которого увеличивается при воспалении, инфекционных и ревматических заболеваниях, некоторых злокачественных процессах, при заместительной терапии эстрогенами и беременности (в III триместре – в 2 раза). В норме печень в сутки секретирует 34 мг/кг массы тела А1АТ, а при развитии воспалительного и опухолевого процесса концентрация увеличивается в 2-5 раз. У пациентов с дефицитом данного белка не происходит его продуцирования. При разделении фракций общего белка в агарозном геле А1АТ находится во фракции альфа‑1-глобулинов.
Причиной поражения ткани легкого при дефиците А1АТ является дисбаланс в системе протеолиз-антипротеолиз. В этом случае происходит неконтролируемое повышение активности протеолитических ферментов, прежде всего нейтрофильной эластазы, в результате чего эластичные волокна и другие структуры экстрацеллюлярного матрикса нижних отделов дыхательных путей подвергаются медленной деструкции. Это приводит к потере эластичности легочной ткани, развитию обструктивных нарушений и эмфиземы.
Генетика А1АТ
А1АТ кодируется геном SERPINA1 (ингибитор сериновой пептидазы, clade A), находящимся в локусе ингибитора протеиназы (Pi) на длинном плече хромосомы 14q32.1 [13]. Локус Pi имеет длину 12,2 kb и состоит из четырех кодирующих (II-V), трех некодирующих (1A, 1B, 1C) экзонов и шести интронов. Экзоны II-V кодируют и содержат информацию о последовательности, которая определяет сам белок. Стартовый кодон (АТГ) служит для трансляции мРНК и сигнального пептида в экзоне II, а стоп-кодон (ТАА) находится в экзоне V. Область, кодирующая ингибиторный метионин реактивного центра (Met358), расположена в пределах V экзона. После транскрипции мРНК А1АТ транслируется на рибосоме, связанной с эндоплазматическим ретикулумом, и продуцирует препротеин с 418 аминокислотами. Сигнальный пептид, содержащий 24 аминокислотных остатка, удаляется во время секреции в эндоплазматический ретикулум, где происходит гликозилирование белка с высоким содержанием маннозных углеводов, и белок приобретает соответствующую конфигурацию шаровидного дерева. Полная сборка белка происходит внутри аппарата Гольджи, откуда происходит его секреция. Ген SERPINA1 является высокополиморфным.
Генетический полиморфизм А1АТ
Кодирующий ген А1АТ SERPINA1 является высокополиморфным с более чем 125 SNP (точечный нуклеотидный полиморфизм). Варианты белка A1AТ классифицируются по системе Pi (Protease inhibitor), и каждый вариант идентифицируется путем миграции при электрофорезе в агарозном геле. Эти различия в миграции связаны с изменениями заряда белка, который зависит от аминокислотного состава [14]. Изоэлектрическая фокусировка в узком диапазоне рН (4,2-4,9) позволила идентифицировать больше вариантов А1АТ по сравнению с электрофорезом в агарозном геле. Аллелям были присвоены символы в соответствии с электрофоретической подвижностью. При этом анодные варианты отмечены первыми, а катодные – последними буквами. Аллель Z (наиболее частый дефицитный вариант) двигается медленно и расположена близко к катоду (Z‑область). М‑аллель (нормальная аллель) продвигается к середине геля и попадает в М‑область [15].
Все варианты А1АТ подразделяются на четыре класса согласно уровню белка в сыворотке и его функциональной активности:
- Нормальные. А1АТ вырабатывается в достаточном количестве и с нормальными свойствами.
- Дефицитные. Вырабатывается недостаточное количество А1АТ. Причиной недостаточности ингибитора протеаз является его внутриклеточное накопление в гепатоцитах, где он синтезируется, или разрушение с высвобождением в кровоток минимальных его количеств.
- Нулевые. А1АТ полностью отсутствует. Недостаточность обусловлена нарушениями транскрипции и синтезом неполноценного или нестабильного белка, разрушающегося еще до секреции.
- Дисфункциональные. Уровень А1АТ не отличается от нормы, но белок не может выполнять свои функции, что обусловлено снижением или утратой антиэластазной активности.
Нормальные варианты А1АТ
Характеризуются достаточным уровнем в сыворотке и нормальной функциональной активностью для ингибирования нейтрофильной эластазы. Самым «распространенным» нормальным вариантом гена Pi является аллель М (более 95%). Среди европеоидов М1 (Val213) встречается наиболее часто, а М1 (Ala213), М2 и М3 – значительно реже. К «редким» нормальным вариантам с частотой встречаемости менее 1% относятся М4, BАlhambra, F, PStAlbans и XChristchurch. Редкие варианты названы по имени места рождения самого старого человека (в родословной) при лабораторном тестировании. Гомозиготные и гетерозиготные PiM характеризуются нормальным уровнем А1АТ в сыворотке (20-50 мкмоль/л) и нормальной функциональной активностью [16]. Необходимо отметить, что именно от М‑субтипов путем мутирования произошли все остальные нормальные и патологические варианты гена Pi.
Дефицитные варианты А1АТ
Низкое содержание А1АТ в сыворотке по сравнению с нормальными вариантами. Было идентифицировано несколько мутаций, связанных с А1АТ. Наиболее распространенными являются Z- и S‑аллели. К редким дефицитным вариантам А1АТ относятся MMalton, MMineralSprings, MNichinan, MProcida, PLowell, SIiyama и др. Оценка возраста вариантов А1АТ, основанная на микросателлитной вариации, показывает, что дефицитная Z‑аллель появилась от 107 до 135 поколений назад, в эпоху неолита. Наибольшая частота вариации Z‑аллели наблюдается у европеоидов, редкая или полностью отсутствует – у азиатов и африканцев [17]. Аллель дефицита PiS имеет возраст от 279 до 479 поколений. Предполагается, что она могла возникнуть на Пиренейском полуострове [18].
Z‑аллель
Вариант Z представляет собой «классический» дефицитный вариант А1АТ, который имеет происхождение после точечной мутации от М1 (Ala213). Результатом мутации является замена нуклеотида GAG, кодирующего аминокислоту Glu342, на нуклеотид AAG, кодирующий Lys342 [19]. Z‑мутация приводит к изменению структуры белка [20].
Процесс полимеризации зависит от концентрации и температуры. Патология в посттрансляционной модификации белка вызывает накопление полимеров А1АТ в эндоплазматическом ретикулуме с резким снижением скорости секреции. Патологический белок накапливается в гепатоцитах и образует тела включения (агрегаты). А1АТ‑полимеры являются токсичными для гепатоцитов и могут вызвать разнообразные повреждения печени, начиная от неонатального гепатита до ювенильного цирроза и гепатоцеллюлярной карциномы у взрослых. Вследствие аккумулирования полимера гепатоциты, гомозиготные PiZZ, секретируют только 10-15%, а гетерозиготный PiZM – 50% от уровня нормальной циркуляции А1АТ. Кроме низкого уровня циркуляции, белок Z также менее эффективно ингибирует эластазу. Поэтому у людей с PiZZ количественные и качественные дефекты А1АТ приводят к ранней стадии ХОБЛ, включая эмфизему и хронический бронхит. Полимеры А1АТ были обнаружены в бронхоальвеолярной лаважной жидкости у гомозигот PiZ с эмфиземой. Это конформационное превращение может способствовать уменьшению уровней функционирования ингибитора протеиназы в легких и, как следствие, усугубить повреждение тканей легкого. Исследования J. Parmar и соавт. [21], A. Mulgrew и соавт. [22] показали, что ZA1AT, локально продуцируемый на эпителиальной поверхности легкого, полимеризуется, и полимеры А1АТ демонстрируют проапоптотические и провоспалительные эффекты. В этих исследованиях обнаружено, что ZА1АТ, в отличие от МА1АТ, является неэффективным антипротеазным ингибитором и может выступать сильным нейтрофильным хемоаттрактантом, что обусловливает появление постоянного источника воспаления в легких у людей с дефицитом А1АТ. Таким образом, полимеризация локального продуцирования ZА1АТ является фактором, способствующим воспалению легких у лиц с дефицитом А1АТ.
S‑аллель
В отличие от аллели Z, аллель S вызывает только умеренную недостаточность А1АТ в сыворотке. Генетическая последовательность S‑варианта образуется из М1 (Val213) в результате мутации, которая вызывает замещение нуклеотида GAA, кодирующего аминокислоту Glu264 на нуклеотид GTA, кодирующий Val264. Одиночная мутация варианта S приводит к спонтанному формированию полимера, но медленнее, чем вариант Z, и не влияет на способность ингибировать эластазу нейтрофилов [23]. Более медленная полимеризация приводит к уменьшению S‑вариантов в печени, поэтому уровни в сыворотке составляют 60% от нормальной М‑аллели. Так, носители аллели S (PiSS, PiSZ и PiMS) имеют 52, 32 и 75% соответственно от нормального уровня. Кроме того, ZA1AT образуют гетерополимеры с SA1AT, которые объясняют случаи цирроза печени у PiSZ‑пациентов.
Нулевые варианты А1АТ
Характеризуются модификацией важной части гена с неопределяемой мРНК. Хотя они являются чрезвычайно редкими, встречаются во всех популяциях. Частота встречаемости среди европеоидов составляет менее 0,1%. Варианты нулевых аллелей обозначаются как Q0, а не Pi. Мутации Null не приводят к секреции белка или образованию полимеров. У пациентов с Null-мутацией отмечается значительное снижение функции легких по сравнению с пациентами, имеющими PiSZ и PiZZ, и они имеют особенно высокий риск развития эмфиземы [24]. Раннее выявление нулевых носителей важно для профилактических и терапевтических вмешательств. Аллель PiNullBellingham отличается от нормального гена М1 (Val213) мутацией в экзоне II, где кодон для Lys217 (AAG) изменяется на стоп-кодон (TAG). У гомозиготных индивидуумов с PiNullBellingham отмечается полное отсутствие А1АТ и эмфизема развивается намного раньше, чем у более распространенной формы PiZZ. Мутация NullIsola di Procida вызвана полным удалением экзонов II-V гена SERPINA1 [25].
Дисфункциональные варианты А1АТ
Белок синтезируется в нормальных количествах, но имеет изменения в функционировании. Аллель PiPittsburgh является мутацией, которая возникает на активном сайте А1АТ и ответственна за изменение функционирования продукта гена. А1АТ становится мощным ингибитором тромбина и фактора XI, а не эластазы, что приводит к развитию кровотечения.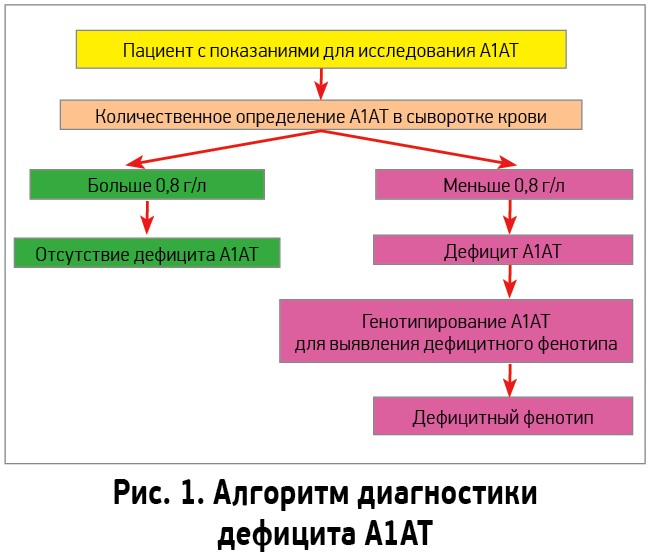
Показаниями для исследования уровня А1АТ в сыворотке крови являются:
- ХОБЛ среди пациентов молодого возраста (курящие и некурящие);
- раннее появление эмфиземы легких;
- бронхоэктатическая болезнь;
- бронхиальная астма, плохо отвечающая на лечение;
- частые обострения бронхита, пневмонии;
- быстрое прогрессивное снижение функции внешнего дыхания;
- хронический кашель и одышка;
- семейный анамнез заболеваний легких и печени;
- заболевание печени неясного генеза;
- наследственный дефицит А1АТ;
- панникулит/васкулит.
 В случае подозрения на дефицит А1АТ при наличии одного или нескольких из перечисленных критериев предлагается использовать алгоритм диагностики, представленный на рисунке 1 [26].
В случае подозрения на дефицит А1АТ при наличии одного или нескольких из перечисленных критериев предлагается использовать алгоритм диагностики, представленный на рисунке 1 [26].
Референтные значения А1АТ в сыворотке
Нормальное содержание А1АТ в сыворотке составляет 0,9-2,0 г/л, или 18,4 (17-37) мкмоль/л, или 100 (90-200) мг/дл. Дефицитом А1АТ считается уровень в сыворотке крови менее 0,8 г/л (15 мкмоль/л, или 80 мг/дл).
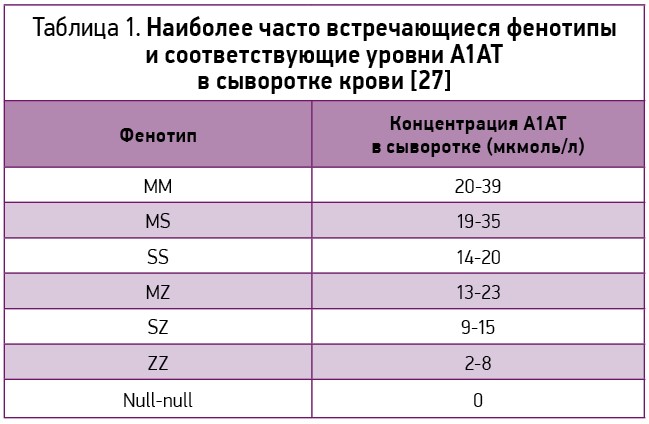
Наиболее часто встречающиеся фенотипы и соответствующие уровни А1АТ в сыворотке представлены в таблице 1.
Для выявления групп риска среди пациентов с первичной эмфиземой легких считается целесообразным проведение скрининга на дефицит А1АТ с последующим его генотипированием (табл. 2).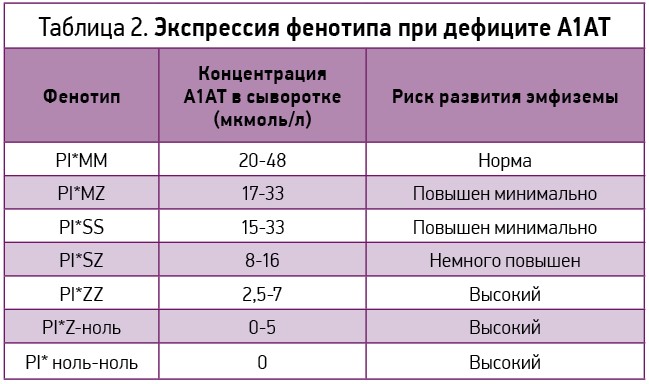
Лабораторные методы оценки дефицита А1АТ
Всем пациентам с подозрением на наследственный дефицит А1АТ рекомендуется измерение уровня данного белка в сыворотке крови. Для количественного определения концентрации А1АТ в сыворотке крови применяются нефелометрические и турбидиметрические тесты. Интерпретируя результаты, следует учитывать, что при инфекционных и воспалительных реакциях, опухолях, стрессе, шоке, беременности, приеме эстрогеносодержащих препаратов уровень А1АТ в крови повышается. Исследование рекомендуется проводить вне периода обострения, связанного с дефицитом А1АТ. К качественным методам определения А1АТ относят генотипирование с помощью молекулярно-биологических технологий и молекулярное фенотипирование. Для анализа фенотипов А1АТ широкое распространение получил метод изоэлектрофокусирования, который позволяет определять более 100 различных фенотипических вариантов А1АТ, не прибегая к дорогостоящему секвенированию полной последовательности гена Pi (рис. 2) [28].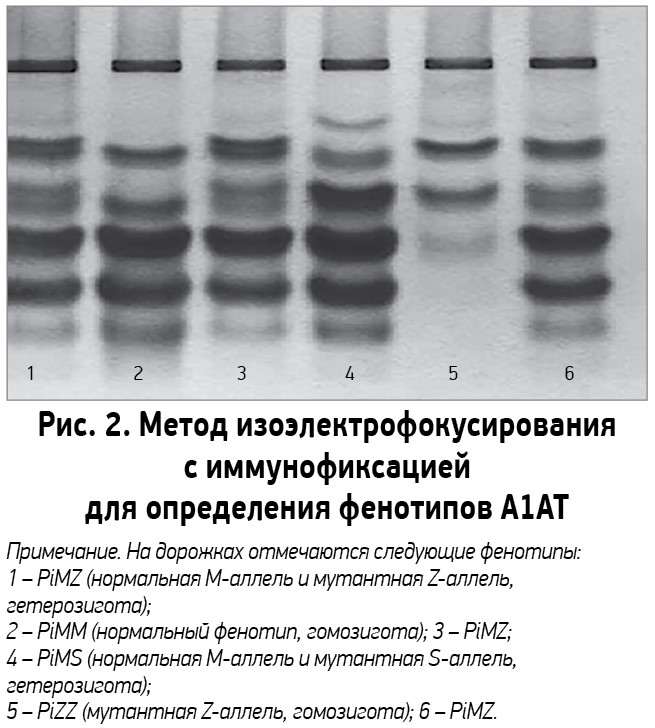
В клинической практике недостаточность А1АТ у 95% пациентов ассоциируется с аллелями PiZ и Pi S.
Выводы
- Дефицит А1АТ является недооцененным заболеванием и относится к редким ввиду неактуальности проведения скрининга в Украине. Главной причиной этого является ограниченность знаний врачей-клиницистов о данном заболевании респираторной системы.
- Часто проходит длительный период времени от начала заболевания до постановки диагноза (5-7 лет).
- В большинстве случаев диагноз недостаточности А1АТ основывается только на определении концентрации данного белка в сыворотке без генетической верификации.
- Данное заболевание часто не распознают или диагностируют неправильно (особенно дефицит PiZZ).
- Тяжесть заболевания определяется не только вариантом мутации, но и внешними факторами окружающей среды, что может провоцировать и отягощать проявление дефицита.
- Для лечения данного заболевания используется А1АТ‑замещающая терапия.
Список литературы находится в редакции.
Медична газета «Здоров’я України 21 сторіччя» № 24 (469), грудень 2019 р.