



Наследственный рак органов желудочно-кишечного тракта
Выделение такого гена означало бы выход из пещеры на свет…
Роберт Вайнберг
.jpg) В предыдущем номере была опубликована статья, посвященная семейному и наследственному раку молочной железы. Сегодня мы продолжаем обсуждать тему наследственных опухолей на примере рака органов желудочно-кишечного тракта (желудка, толстой кишки и поджелудочной железы).
В предыдущем номере была опубликована статья, посвященная семейному и наследственному раку молочной железы. Сегодня мы продолжаем обсуждать тему наследственных опухолей на примере рака органов желудочно-кишечного тракта (желудка, толстой кишки и поджелудочной железы).
Наследственный рак желудка
Сегодня принято считать, что наследственный рак составляет не менее 10% от всех злокачественных опухолей желудка.
Известно, что от наследственного рака желудка умер Наполеон Бонапарт, его отец Карл Бонапарт и родная сестра Мария Бонапарт (Боргезе). Предрасположенность к наследственному раку желудка передается геном CDH1, который кодирует белок E-cadherin. Этот белок участвует в поддержании эпителиальной архитектуры тканей и сохраняет клеточную адгезию.
Мутации в CDH1 ассоциированы с развитием особого типа карциномы – диффузного перстневидно-клеточного рака желудка (linitis plastica), пожизненный риск возникновения которого при наличии генетической аномалии у мужчин составляет 70%, у женщин – 55%. При усеченном (truncated) типе гена CDH1 у женщин также имеется риск развития долькового рака молочной железы (РМЖ) на уровне 40%, о чем было подробно написано в предыдущей статье. Риск развития рака другой локализации при этой мутации неясен.
Роль мутации CDH1 в передаче предрасположенности к наследственному раку желудка хорошо видна на следующем клиническом примере, который приведен в материалах образовательного портала Европейского общества медицинской онкологии (ESMO).
У 35-летней женщины диагностирован клинически ранний дольковый рак молочной железы (pT1cpN0G2 ER+, PgR+, HER2-, Ki67 – 17%). Анамнез рака в этой семье отягощен: мать заболела и умерла от РМЖ в 43 года, дядя по материнской линии умер в 37 лет от рака желудка, бабушка также умерла от рака желудка в возрасте 50 лет.
В связи с молодым возрастом начала заболевания и семейной историей рака женщине было проведено генетическое тестирование. Обнаружена мутация CDH1 и отсутствие мутаций BRAF1/2. Это позволило исключить у нее наследственный BRAF-ассоциированный тип РМЖ. Пациентке выполнена радикальная мастэктомия и назначено адъювантное лечение агонистами гонадотропин-рилизинг-гормона и тамоксифеном в течение 5 лет. Из-за наличия мутации CDH1 на протяжении 3 лет после мастэктомии женщине ежегодно проводили скрининговую эндоскопию и биопсию желудка с целью обнаружения возможной карциномы. Во всех случаях результат был отрицательным.
Несмотря на полное отсутствие жалоб и отрицательные результаты обследования, женщина сама настояла на выполнении ей профилактической тотальной гастрэктомии. При послеоперационном гистологическом исследовании в удаленном желудке был обнаружен инвазивный перстневидно-клеточный рак стадии IA, pT1cpN0G2.
Родным братьям пациентки провели молекулярное тестирование, оба оказались носителями мутации CDH1.
Второму 30-летнему брату также провели гастроскопию, однако результаты биопсии оказалась отрицательными. Несмотря на это пациент настоял на выполнении профилактической тотальной гастрэктомии. В послеоперационном материале удаленного желудка при гистологическом исследовании признаков опухоли не обнаружено.
Данное наблюдение не только демонстрирует современные возможности профилактической онкологии, основанной на проведении молекулярного профилирования генов наследственного рака, но и ставит ряд конкретных клинических вопросов.
? Кому показано проведение генетического тестирования при подозрении на наследственный рак желудка?
Обязательное тестирование (включая родственников 1-й и 2-й степени родства) проводят в семьях, в которых имеется следующий анамнез:
- наличие двух случаев рака желудка в семье, независимо от возраста заболевших (один подтвержденный диффузный рак желудка);
- один случай диффузного рака желудка в возрасте младше 40 лет;
- один случай диффузного рака желудка и долькового РМЖ у одного члена семьи в возрасте младше 50 лет.
Семьи, в которых крайне желательно провести тестирование для определения риска развития наследственного рака желудка:
- случай двустороннего долькового РМЖ у одного члена семьи;
- два или более случаев долькового РМЖ в семье в возрасте младше 50 лет;
- сочетание диффузного рака желудка и незаращения губы/неба у одного члена семьи;
- выявление перстневидных клеток in situ при гастробиопсии.
? С какого члена семьи нужно начинать генетическое тестирование?
Генетические тесты в семье нужно начинать с заболевшего. Не следует проводить профилактические исследования у всех здоровых членов семьи. Если мутация гена у заболевшего будет найдена, тестированию должны подвергаться все родственники 1-й линии.
Если мутация гена не будет выявлена, дальнейшее тестирование в семье не проводится. В этом случае рак желудка следует расценивать как семейный без характерной известной мутации.
? Нужно ли выполнять профилактическую гастрэктомию всем членам семьи, у которых есть мутация CDH1, или достаточно ежегодно проводить серийную гастробиопсию?
Опыт свидетельствует, что проведение диагностической гастроскопии с биопсией внешне здоровой слизистой оболочки может позволить обнаружить раковые клетки в очень малом проценте случаев.
Внутрислизистая перстневидно-клеточная инвазивная карцинома или карцинома in situ может располагаться на крайне ограниченных участках слизистой оболочки между нормальными неизмененными клетками (рис. 1).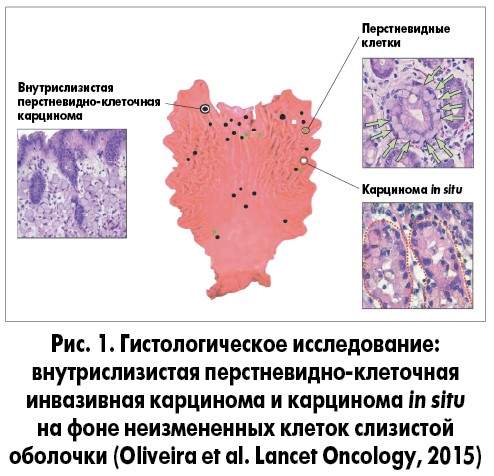
Строгой рекомендацией при обнаружении мутации CDH1 у здорового человека с наличием рака в семейном анамнезе является выполнение полной гастрэктомии, а не проведение скрининговой гастробиопсии.
? Какие рекомендации должны получить пациенты после выполнения профилактической гастрэктомии?
Поскольку мутация CDH1 в 40% случаев ассоциирована с дольковым РМЖ и в 5% – с раком ободочной кишки, пациенты, перенесшие профилактическую гастрэктомию, должны проходить скрининг РМЖ с 30 лет и скрининг рака толстой кишки с 40 лет.
? Кому показано генетические консультирование при подозрении на наследственный рак желудка?
Перед молекулярным исследованием всем пациентам необходимо провести генетическое консультирование. Оно показано следующим категориям:
- пациентам младше 40 лет с установленным диагнозом диффузный рак желудка;
- лицам, имеющим в семье родственников с диффузным раком желудка;
- пациентам, имеющим в семье родственников младше 50 лет с дольковым РМЖ.
Другие генетические маркеры наследственного рака желудка
CDH1 – основной ген предрасположенности к наследственному раку желудка, однако существуют и другие редкие генетические аномалии. Описаны несколько семей с инактивацией гена CTNNA1, семьи с сочетанием аденокарциномы и проксимального полипоза желудка (GAPPS), семейный рак желудка кишечного типа (FIGC), причины которого не выявлены.
Клиническое значение имеет также атипичный рак желудка при других наследственных генетических синдромах – ювенильного полипоза (мутация SMAD4), Пейтца – Егерса (мутация STK11), Ли – Фраумени (мутация TP53).
Синдром Линча
 Генри Линч, известный онколог из Небраски, описал многочисленные случаи рака в нескольких поколениях одной семьи. Члены этой семьи болели колоректальным раком (КРР), раком эндометрия, яичника, желудка, тонкой кишки, мочевых путей, поджелудочной железы, кожных желез и опухолями головного мозга. Этот опухолевый синдром был назван синдром Линча.
Генри Линч, известный онколог из Небраски, описал многочисленные случаи рака в нескольких поколениях одной семьи. Члены этой семьи болели колоректальным раком (КРР), раком эндометрия, яичника, желудка, тонкой кишки, мочевых путей, поджелудочной железы, кожных желез и опухолями головного мозга. Этот опухолевый синдром был назван синдром Линча.
Клинически синдром Линча можно заподозрить в следующих случаях:
- в семье есть несколько близких родственников моложе 70 лет с раком ободочной кишки;
- КРР возникает в возрасте до 40 лет;
- у одного человека имеется синхронный первично-множественный рак ободочной кишки.
При развитии рака кишечника до 40 лет вероятность наличия у больного синдрома Линча повышается до 20%, при развитии рака кишечника до 70 лет вероятность синдрома Линча невелика (1-2%). У 35% пациентов синдром Линча может быть заподозрен клинически на основании семейного анамнеза.
Молекулярной основой синдрома является микросателлитная нестабильность (MSI). Ее можно выявить с помощью иммуногистохимического исследования опухолевой ткани. Согласно обновленным рекомендациям Национальной онкологической сети США (2017), во всех случаях метастатического рака ободочной кишки должны быть проведены исследования на дефицит MMR и наличие MSI.
У больных с неметастатическим раком толстой кишки тест MSI следует проводить согласно рекомендациям Bethesda:
- если КРР диагностирован в возрасте до 50 лет;
- при наличии синхронных или метахронных опухолей в кишечнике;
- при наличии рака кишечника и сопутствующих внекишечных опухолей, независимо от возраста;
- если КРР диагностирован у пациента, у которого ранее в возрасте до 50 лет был выявлен внекишечный рак;
- если КРР диагностирован у пациента, у которого ранее, независимо от возраста, были обнаружены 1 или 2 внекишечные опухоли.
Опухоли при синдроме Линча ассоциированы с мутациями зародышевой линии в генах MLH1, MSH2, MSH6, EPCAM и PMS2. Распространенность таких мутаций в общей популяции составляет 0,4%.
Риск развития рака отличается в зависимости от мутации различных генов. Пожизненный риск при мутации PMS2 повышен на 15-20%, MSH6 – на 30-70%, MSH2 – на 40-80%, при мутации MLH1 – на 40-80%. Чаще всего при таких аномалиях развивается КРР (60-70%) и рак эндометрия (30-70%).
Доля синдрома Линча составляет 5% от всех случаев рака ободочной кишки, он имеет свои особенности. Обычно синдром Линча возникает в молодом возрасте (в среднем до 40-45 лет), характеризуется быстрым прогрессированием от аденомы до рака, первично-множественным синхронным и метахронным поражением и сочетанием с экстраколитическим раком (в первую очередь раком эндометрия).
Постановка диагноза синдром Линча важна как для пациента, поскольку это влияет на тактику лечения, так и для его наследников, поскольку с помощью профилактических мероприятий можно предотвратить развитие опухоли.
Тактика лечения при синдроме Линча может быть продемонстрирована на примере следующего клинического случая.
У 27-летней женщины был диагностирован рак правой половины ободочной кишки с метастазами в печени и брюшине – T3N2M1 (hep, per), аденокарцинома G2. Имеется отягощенный семейный анамнез – мать и бабушка умерли от рака кишечника и рака эндометрия. Учитывая молодой возраст заболевшей и семейный анамнез рака, было проведено молекулярное профилирование. Обнаружена germline MLH1 мутация и MSI+, что позволило установить диагноз синдрома Линча. 7-летняя дочь больной также оказалась носителем мутации MLH1.
Из-за распространенности опухолевого процесса хирургическое лечение не было показано. Стандартные химио- и таргетная терапия были неэффективны. Учитывая наличие MSI+, больной была назначена анти-PD-L1 иммунотерапия (основание – результаты исследований KEYNOTE‑164, KEYNOTE‑158 и рекомендации Управления по контролю качества пищевых продуктов и лекарственных препаратов США), что позволило контролировать течение заболевания без прогрессирования на протяжении 2,5 года.
? Какие рекомендации должна получить дочь, унаследовавшая патологическую мутацию MLH1?
Клинически рак при синдроме Линча выявляют после достижения репродуктивного возраста. Носители мутации должны получить следующие рекомендации:
- проведение колоноскопии каждые 1-2 года, начиная с 20-25 лет;
- проведение биопсии эндометрия каждые 1-2 года, начиная с 30-35 (определение CA‑125 и выполнение трансвагинального ультразвукового исследования неэффективно);
- гастроскопия рекомендована в семьях с высокой распространенностью опухолей желудка или двенадцатиперстной кишки;
- для носителей мутации MLH1 или MSH2 рекомендована профилактическая гистерэктомия и овариэктомия в возрасте после 35 лет (если деторождение завершено);
- профилактическая колонэктомия при синдроме Линча не выполняется.
Наследственный полипоз кишечника
 Наследственный полипозный синдром толстой кишки выявляют у 5% больных с опухолями ободочной кишки. В 2001 г. с этим синдромом ассоциировалась только одна мутация, ответственная за формирование наследственного рака, – АРС. За последние 15 лет количество мутаций, ответственных за развитие полипозного рака кишечника, резко увеличилось, что позволило создать молекулярную классификацию этого синдрома.
Наследственный полипозный синдром толстой кишки выявляют у 5% больных с опухолями ободочной кишки. В 2001 г. с этим синдромом ассоциировалась только одна мутация, ответственная за формирование наследственного рака, – АРС. За последние 15 лет количество мутаций, ответственных за развитие полипозного рака кишечника, резко увеличилось, что позволило создать молекулярную классификацию этого синдрома.
Клинически принято различать аденоматозный полипоз кишечника (при наличии более 10 синхронных аденом), синдром Пейтца – Егерса (полипы желудка и кишечника), синдром ювенильного полипоза (более 5 ювенильных полипов), синдром зубчатого полипоза (20‑30 зубчатых полипов).
Полипы кишечника (рис. 3) имеют разное морфологическое строение, частоту малигнизации, риск развития колоректальной карциномы и других злокачественных опухолей. При синдроме наследственного семейного полипоза повышен риск возникновения не только КРР, но и рака щитовидной железы, печени, десмоида и медуллобластомы. При синдроме Пейтца – Егерса часто развиваются рак желудка, двенадцатиперстной, тонкой кишки, яичника, шейки матки и яичка. Синдром ювенильного полипоза часто сочетается с КРР, раком желудка и тонкой кишки.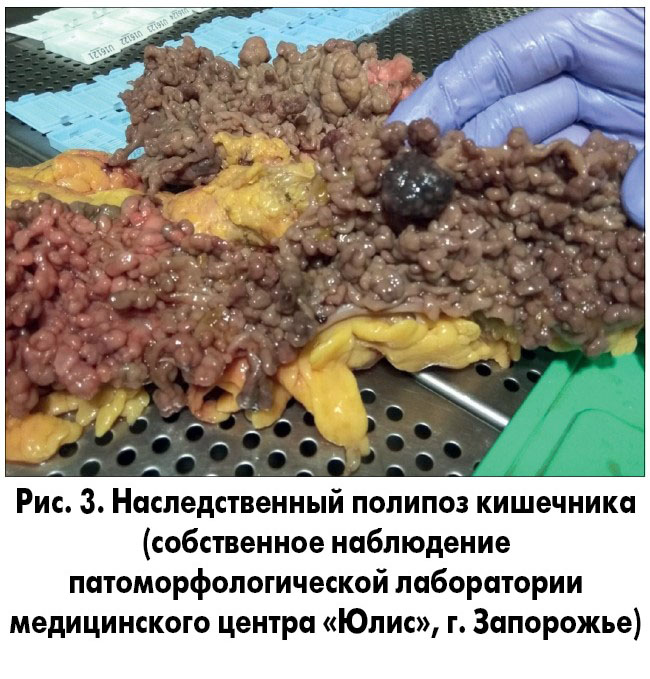
Для некоторых синдромов наследственного кишечного полипоза характерны многочисленные внекишечные проявления: отставание в развитии, макроцефалия, остеомы, липомы, поражения кожи, пигментные изменения, разрастание десен, сосудистые мальформации, нарушение развития ногтей.
Из-за риска формирования опухолей кишечника пациентам с анамнезом и мутациями, ответственными за развитие наследственного полипоза, следует придерживаться следующих рекомендаций:
- проведение сигмоидоскопии следует начать в возрасте 12-14 лет;
- если аденомы не обнаружены, у носителей мутаций эндоскопию выполняют пожизненно;
- если характерные мутации не выявлены, наблюдение осуществляется с 40-летнего возраста каждые 2 года, позже – каждые 3-5 лет;
- если обнаружен аденоматоз кишечника, следует рекомендовать колэктомию.
Наблюдение за пациентами с анамнезом и мутациями, ответственными за развитие наследственного полипоза, должно проводиться также из-за риска развития внекишечных опухолей. Рекомендации включают:
- выполнение гастроскопии с 20-25 лет каждые 5 лет (цель – обнаружение полипов желудка);
- проведение ультразвукового исследования шеи для скрининга аденом щитовидной железы;
- компьютерная томография брюшной полости – регулярно (диагностика десмоидных опухолей);
- капсульная эндоскопия – выявление аденом тонкой кишки.
Наследственный рак поджелудочной железы
Генетическими факторами риска развития рака поджелудочной железы (РПЖ) являются мутации BRCA1,2, P16, ATM, STK11, PRSS1/PRSS2, SPINK1, PALB2. Наиболее частой причиной наследственного РПЖ является мутация в гене BRCA2. Обнаружены множественные генетические полиморфизмы – ABO, NR5A2, TERT, Pdx1, BCAR1, CTRB1, CTRB2, ZNRF3, LINC00673, ETAA1, TP63, SUGCT. Приблизительно 10% пациентов с РПЖ имеют семейную историю рака. В 80% семей с анамнезом РПЖ генетические аномалии не выявлены.
РПЖ может быть частью некоторых наследственных синдромов. Наследственные синдромы, связанные с повышенным риском этого вида рака:
- наследственный синдром рака молочной железы и рака яичника;
- синдром семейной атипичной множественной меланомы;
- синдром Линча;
- семейный аденоматозный полипоз;
- синдром атаксии-телеангиэктазии;
- синдром Пейтца – Егерса;
- синдром наследственного панкреатита.
Самый высокий риск развития наследственного РПЖ имеют больные с синдромом Пейтца – Егерса (до 40%) и синдромом наследственного панкреатита (до 50%). К группе высокого риска возникновения наследственного РПЖ относятся следующие категории пациентов:
- лица с тремя или более кровными родственниками с РПЖ, по крайней мере с одним родственником
1-й степени; - лица как минимум с двумя родственниками 1-й степени с РПЖ;
- пациенты с синдромом Пейтца – Егерса, независимо от семейного анамнеза РПЖ;
- носители мутации CDKN2A/p16 с одним больным родственником 1-й степени;
- носители мутации BRCA2 с одним больным родственником 1-й степени;
- носители мутации PALB2 с одним больным родственником 1-й степени;
- носители MMR с синдром Линча и одним больным родственником 1-й степени.
Следует учитывать следующие рекомендации по наблюдению за пациентами с высоким риском развития наследственного РПЖ:
- необходимо ежегодно проводить эндоскопическую ультрасонографию и/или магнитно-резонансную томографию;
- наблюдение следует начинать в возрасте 50 лет (или на 10 лет раньше, чем возраст самого младшего заболевшего родственника);
- у пациентов с наследственным панкреатитом или синдромом Пейтца – Егерса наблюдение начинают с 30 и 40 лет;
- у носителей мутаций профилактическая панкреатэктомия не выполняется (консенсус не достигнут);
- нет убедительных доказательств того, что скрининг связан с уменьшением заболеваемости и смертности от РПЖ.
Таким образом, наследственный рак органов желудочно-кишечного тракта представляет сложную гетерогенную группу онкологических заболеваний с характерной клинической картиной и прогнозом. Особенностью наследственного рака является риск развития метахронной опухоли в том же или другом органе.
Рекомендации по ведению пациентов с наследственным раком должны быть ориентированы не только на онкологических больных, но и на их родственников – здоровых носителей патологической мутации, что позволит предотвратить у них развитие наследственной злокачественной опухоли.
Современные технологии выявления генов наследственного рака, основанные на изучении полноценных панелей, а не одной мутации или SNP, позволяют с высокой достоверностью установить уровень риска развития заболевания.
В Украине следует более активно развивать программы генетического скрининга наследственного рака.
Тематичний номер «Онкологія, Гематологія, Хіміотерапія» № 3 (64) 2020 р.
СТАТТІ ЗА ТЕМОЮ Гастроентерологія
Метаболічноасоційована жирова хвороба печінки (МАЖХП) є однією з найактуальніших проблем сучасної гепатології та внутрішньої медицини в цілому. Стрімке зростання поширеності ожиріння та цукрового діабету (ЦД) 2 типу в популяції призвело до істотного збільшення кількості хворих на МАЖХП, яка охоплює спектр патологічних станів від неускладненого стеатозу до алкогольної хвороби печінки та цирозу, що розвиваються на тлі надлишкового нагромадження ліпідів у гепатоцитах. ...
Інфекція Helicobacter pylori (H. pylori) офіційно визнана інфекційним захворюванням і включена до Міжнародної класифікації хвороб (МКХ) 11-го перегляду, тому рекомендовано лікувати всіх інфікованих пацієнтів. Проте, зважаючи на широкий спектр клінічних проявів, пов’язаних із гастритом, викликаним H. pylori, лишаються специфічні проблеми, які потребують регулярного перегляду для оптимізації лікування. ...
Гостра лімфобластна лейкемія (ГЛЛ) є найпоширенішим онкогематологічним захворюванням у дітей і складає значну частку серед лейкемій у дорослих. Незважаючи на значні успіхи в лікуванні ГЛЛ у дітей, де рівень виліковності сягає 90%, результати терапії у дорослих залишаються незадовільними. У рамках науково-практичної конференції з міжнародною участю «Діагностика та лікування гематологічних захворювань: підведення підсумків 2023 року» (15-16 грудня 2023 року) проведено секцію, присвячену ГЛЛ....
Відтворення майбутнього здорової нації – один з найважливіших сенсів існування теперішнього покоління. День боротьби з ожирінням нагадує нам про поширеність цього проблемного явища і важливість попередження його наслідків. Ожиріння може мати вплив на різні аспекти здоров'я, включаючи репродуктивне....