



Мукополісахаридоз І типу – рідкісна причина затримки росту в дітей
Прояви ендокринних та метаболічних розладів, особливо в дитячому віці, спонукають хворих та їхніх батьків звернутися до лікаря практично будь-якої спеціальності. Лише настороженість щодо найпоширеніших захворювань ендокринної системи та міжгалузева кооперація вузьких спеціалістів здатні забезпечити швидке виявлення та оптимальне лікування таких пацієнтів.
 29 травня педіатри, ендокринологи та лікарі суміжних спеціальностей отримали можливість поглибити свої знання в області дитячої ендокринології, узявши участь у конференції «Сучасна дитяча ендокринологія», яка відбулася в уже звичному для більшості онлайн-форматі. Цей захід, добре відомий медичній спільноті, відбувається вже 12-й раз поспіль і залучає не лише вітчизняних, але й іноземних спікерів. Усі доповідачі акцентували увагу на практичних аспектах діагностики та лікування найбільш поширених ендокринних захворювань, базуючися на найновітніших рекомендаціях міжнародних спілок та результатах контрольованих клінічних досліджень. Темою однієї з доповідей, яку було представлено увазі учасників конференції завідувачем кафедри госпітальної педіатрії Запорізького державного медичного університету, доктором медичних наук, професором Геннадієм Олександровичем Леженко, став мукополісахаридоз (МПС) – рідкісне генетичне захворювання і часто недооцінювана причина затримки росту в дітей.
29 травня педіатри, ендокринологи та лікарі суміжних спеціальностей отримали можливість поглибити свої знання в області дитячої ендокринології, узявши участь у конференції «Сучасна дитяча ендокринологія», яка відбулася в уже звичному для більшості онлайн-форматі. Цей захід, добре відомий медичній спільноті, відбувається вже 12-й раз поспіль і залучає не лише вітчизняних, але й іноземних спікерів. Усі доповідачі акцентували увагу на практичних аспектах діагностики та лікування найбільш поширених ендокринних захворювань, базуючися на найновітніших рекомендаціях міжнародних спілок та результатах контрольованих клінічних досліджень. Темою однієї з доповідей, яку було представлено увазі учасників конференції завідувачем кафедри госпітальної педіатрії Запорізького державного медичного університету, доктором медичних наук, професором Геннадієм Олександровичем Леженко, став мукополісахаридоз (МПС) – рідкісне генетичне захворювання і часто недооцінювана причина затримки росту в дітей.
– Захворювання, які спостерігаються з частотою меншою ніж 1:2000, називають рідкісними або орфанними. На сьогодні описано приблизно 7 тис таких захворювань, з яких лише 5% піддаються лікуванню. Особливості перебігу орфанних захворювань (Krzysztof Borski et al., 2017):
- рання маніфестація – 75% починається в дитячому віці;
- 65% хворих мають тяжкий перебіг, що призводить до інвалідизації;
- 50% пацієнтів мають поганий прогноз для життя;
- 35% хворих помирають протягом першого року життя;
- кожен 5-й пацієнт має хронічний больовий синдром.
Багато лікарів не мають настороженості щодо виявлення рідкісних захворювань у своїх пацієнтів, оскільки вважають, що шанс зіткнутися у своїй практиці з такими пацієнтами мізерно малий. Однак вони помиляються. Згідно з результатами пренатального скринінгу рідкісних хвороб накопичення, їх частота вдвічі вища, ніж фактично діагностується (Therrell B.L. et al., 2015).
Одним із таких захворювань є МПС І типу, уперше описаний ще в 1919 році. Причиною захворювання є недостатність ферменту α-L-ідуронідази (IDUA) і накопичення глікозаміногліканів (ГАГ) у тканинах. Захворювання виявляють із частотою 1:100 000, однак в окремій популяції «ірландських мандрівників» частота МПС набагато вища і складає 1:371, а частота носіїв мутантного гена – 1:10 (Meikle P.J. et al., 1999).
Як і інші хвороби накопичення, МПС І типу характеризується ураженням усіх органів та систем. Клінічно це проявляється:
- помутнінням рогівки;
- огрубінням рис обличчя;
- хронічним ринітом і отитами;
- збільшенням язика;
- патологією серцево-судинної системи;
- гепатоспленомегалією;
- пупковими та паховими грижами;
- патологією опорно-рухового апарату: вальгусною деформацією колінних суглобів, кіфозною деформацією хребта та розвитком горба, болями в суглобах та контрактурами, карпальним тунельним синдромом;
- обструкціями дихальних шляхів;
- мікросомією, гідроцефалією;
- хронічною діареєю.
Скарги, зумовлені цими проявами, змушують батьків звертатись і до сімейних лікарів та педіатрів, і до вузьких спеціалістів, тому лікарям із різних сфер медицини треба мати уявлення щодо симптомів і особливостей перебігу МПС.
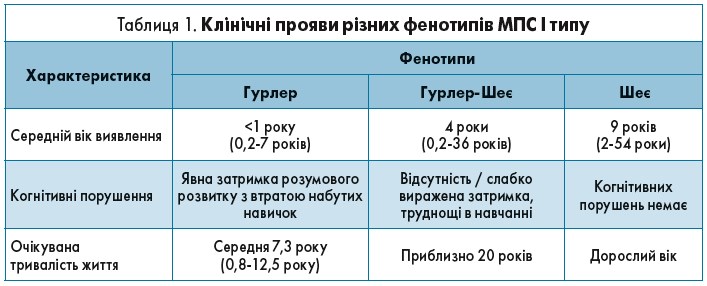
Спектр клінічних проявів МПС залежить від фенотипу захворювання, основні характеристики яких описані в таблиці 1.
МПС І типу, варіанти Шеє та Гурлер- Шеє протікають доволі м’яко, а тому й запідозрити їх важче. Для кращого розуміння алгоритму виявлення цих пацієнтів слід розглянути клінічний випадок.
Клінічний випадок
Батьки пацієнтки Ф., 1999 року народження, звернулися до педіатра (дівчинці на той момент було 4 роки) зі скаргами на незначну скутість суглобів кисті та зміну рис обличчя, відставання в рості. Об’єктивно виявлялися доволі неспецифічні симптоми: зріст 100 см (нижче 75 перцентиля), гепатоспленомегалія та помутніння рогівки. Пацієнтка була проконсультована ендокринологом, кардіоревматологом і генетиком.
Результати інструментальних обстежень
Ехокардіографія: помірна мітральна недостатність із потовщеними клапанами й пролапсом переднього краю; помірне потовщення лівого передсердя і підвищений легеневий тиск.
Рентгенографія: ознаки множинного дизостозу – гіпоплазія кульшового суглобу, яйцеподібна форма поперекових тіл хребців, короткі й широкі п’ясні кістки.
На основі цих даних у пацієнтки був запідозрений МПС. Для підтвердження діагнозу визначили рівень глікозамінгліканів у сечі, який виявився підвищеним, після чого за допомогою ДНК-діагностики виявили гетерозиготну мутацію α-L-ідуронідази W402X/L535F.
У результаті пацієнтці було встановлено діагноз: мукополісахаридоз І типу, Гурлер-Шеє.
Описані в цьому клінічному випадку симптоми мали б змусити лікаря запідозрити різноманітний спектр ендокринних патологій, зокрема порушення, пов’язані з соматотропним гормоном, чи гіпотиреоз. Лише знання симптоматики може наштовхнути лікаря на думку про МПС І типу.
Маркерні симптоми МПС І типу, зі скаргами на які батьки хворої дитини найчастіше звертаються до лікаря (Colville G.A. et al., 1996):
- Непропорційна затримка росту, яка починається з 2 років При цьому рівень гормону росту залишається в нормі.
- Грубі риси обличчя, а при тяжкому перебігу (Гурлер синдром) – прогресуючий лицьовий дисморфізм. Для таких дітей характерне густе жирне й ламке волосся, виступаючий вперед лоб, низько розташовані брови, гіпертелоризм, широкий, плоский, сідлоподібний ніс зі зміщеними вперед ніздрями, макроглосія, прогнатизм та коротка шия.
- Ураження очей, які проявляються у вигляді помутніння рогівки або мегакорнеа, що постійно прогресують, призводять до світлобоязні та неконтрольованого зниження гостроти зору. Крім того, при інфільтрації глікозаміногліканами трабекул розвивається глаукома, а зорового нерва – дегенеративна ретинопатія та атрофія зорового нерва.
- Порушення з боку дихальної системи, зумовлені звуженням дихальних шляхів унаслідок гіпертрофії слизової оболонки, кіфозом і зменшенням рухливості грудної клітки та розвитком рестриктивних порушень через інфільтрацію інтерстиціальної тканини легень. Ці патогенні зміни зумовлюють гучне дихання, зміни голосу, обструкції, рецидивні інфекції, апное уві сні, що призводить до гіпоксії мозку та погіршення неврологічних симптомів. Саме респіраторні ускладнення є основною причиною смерті хворих на МПС І типу (Berger K.I. et al., 2013).
- Ураження кісток та суглобів, які при синдромі Шеє проявляються у вигляді кіфозу, сколіозу, рідше гіперлордозу, але менш виражених, ніж при Гурлер синдромі, дисплазіїю стегна (coxa valga) або западини та контрактури великих суглобів. Для пацієнтів також характерні деформації кінцівок: кистей – так звана «пташина лапа», пальців – так званий «спусковий гачок», колінного суглоба, стопи й великого пальця стопи за типом Genu valgum, pes varus, hallux valgus (Kimberly Morishita et al., 2011).
Зазвичай для встановлення діагнозу МПС І типу необхідна консультація кількох спеціалістів. Діагноз встановлюється на основі характерної клінічної картини, а інколи на визначенні рівня глікозамінгліканів у сечі, однак результати цього обстеження часто бувають хибно негативними.
Підтвердження діагнозу МПС І типу проводиться за допомогою ферментного аналізу активності IDUA в лейкоцитах, плазмі, сироватці або сухих плямах крові (Muenzer J. et al., 2009). Діагностика за методом сухої плями безкоштовно надається ТОВ «Санофі-Авентіс Україна».
Для отримання карт для забору біоматеріалу достатньо звернутися до представників компанії Ярослава Загоруя (тел. 050-358-42-14) або Ігора Нагребецького (тел. 050-382-37-84).
Оптимальне лікування МПС І типу включає в себе як симптоматичні методи та лікарські засоби (видалення гриж, тонзилектомія, ортопедична хірургія), так і специфічну патогенетичну терапію, яка полягає в ферментзамісній терапії (ФЗТ) та трансплантації гемопоетичних стовбурових клітин (de Ru M.H. et al., 2011).
Ключовим фактором, що впливає на прогноз та якість життя пацієнтів, є час виявлення захворювання, адже чим раніше це станеться, тим раніше почнеться замісна терапія, а отже – сформується менше незворотних ускладнень.
Метою ФЗТ є відновлення рівня ензиматичної активності, достатньої для гідролізу накопичених ГАГ і запобігання їх подальшому накопиченню. Пацієнтам призначають препарат ларонідазу (Альдуразим®*), який був синтезований за допомогою технології рекомбінантної ДНК та вперше зареєстрований у світі у 2003 р. (Martins A.M. et al., 2009).
Для наочного розуміння того, наскільки раннє призначення препарату Альдуразим® може вплинути на якість життя пацієнтів, варто розглянути клінічний випадок МПС І типу в брата й сестри (описаної вище пацієнтки Ф.).
Брату й сестрі на сьогодні 13 і 17 років відповідно. Сестрі діагностували МПС I типу у віці 5 років, тоді вона мала класичні риси ослабленої форми хвороби. Після встановлення діагнозу сестрі її брату такий самий діагноз було встановлено невдовзі після народження. І брат, і сестра є носіями складних гетерозиготних IDUA (W402X і L535F)-мутацій. В обох дітей щороку проводили повні клінічні та біохімічні дослідження. У кожної дитини розпочали щотижневі інфузії ларонідази в дозі 0,5 мг/кг – у брата у віці 5 міс, у сестри – у 5 років. Інфузії виконували в лікарні з використанням імплантованих пристроїв венозного доступу; вони добре переносилися пацієнтами, без зареєстрованих побічних реакцій; крім того, було пропущено лише кілька інфузій. Хронологія спостережень та клінічні наслідки в пацієнтів відображені в таблиці 2.
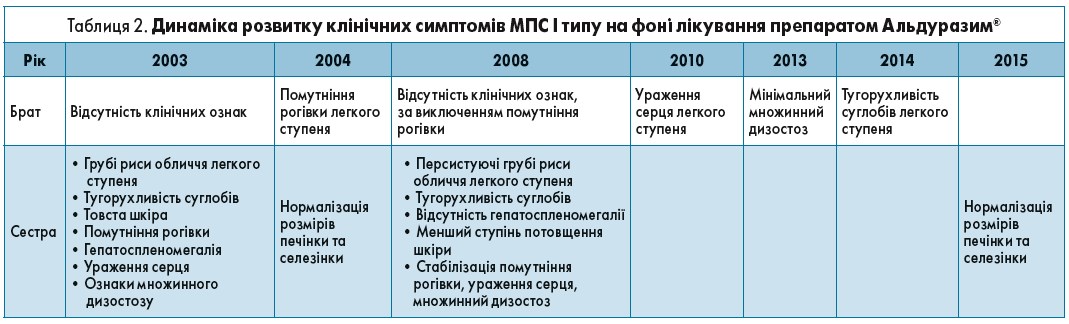
Отже, застосування препарату Альдуразим® у віці до 1 року мінімізувало всі прояви захворювання та забезпечило високу якість життя брата, у той час як початок терапії сестри у віці після 5 років хоч і зупинив подальше прогресування МПС І типу, але лише частково зміг вплинути на регресування сформованих симптомів.
У директиві EC UE/2000 сказано, що пацієнти, які страждають на рідкісні захворювання, мають таке ж право на ефективну терапію, як і пацієнти з більш поширеними патологіями. Саме тому лікування МПС І типу в нашій країні відбувається за державні кошти. Аби дати пацієнтам із МПС І типу шанс на нормальне, якісне життя, лікарям достатньо пам’ятати про це захворювання та якомога раніше проводити діагностику всіх підозрілих випадків.
* Лікарський засіб Альдуразим®, концентрат для розчину для інфузій, 100 Од/мл, № 1. Зареєстрований в Україні. Р.П. UA/8093/01/01. Наказ МОЗ від 03.08.2018 №1449, зміни внесено наказом МОЗ від 31.10.2019 №2205.
Література
- Krzysztof Borski. Characteristics of rare diseases. Режим доступу: https://www.oatext.com/Characteristics-of-rare-diseases.php#gsc.tab=0
- Therrell B.L., Padilla C.D., Loeber J.G. et al. Current status of newborn screening worldwide: 2015. Semin Perinatol. 2015;39(3):171-187. doi:10.1053/j.semperi.2015.03.002. Режим доступу: https://pubmed.ncbi.nlm.nih.gov/25979780/
- Meikle P.J., Hopwood J.J., Clague A.E., Carey W.F. Prevalence of lysosomal storage disorders. JAMA. 1999;281(3):249-254. doi:10.1001/jama.281.3.249. Режим доступу: https://pubmed.ncbi.nlm.nih.gov/9918480/
- Colville G.A., Bax M.A. Early presentation in the mucopolysaccharide disorders. Child Care Health Dev. 1996;22(1):31-36. Режим доступу: https://pubmed.ncbi.nlm.nih.gov/8640962/
- Berger K.I., Fagondes S.C., Giugliani R., Hardy K.A., Lee K.S. et al. (2013). Respiratory and sleep disorders in mucopolysaccharidosis. Journal of inherited metabolic disease, 36(2), 201-210. Режим доступу: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3590419/
- Kimberly M., Ross E. Petty, Musculoskeletal manifestations of mucopolysaccharidoses, Rheumatology, Volume 50, Issue suppl_5, 1 December 2011. P. 19-25. Режим доступу: https://academic.oup.com/rheumatology/article/50/suppl_5/v19/1780256
- Muenzer J., Wraith J.E., Clarke L.A. International Consensus Panel on Management and Treatment of Mucopolysaccharidosis I. Mucopolysaccharidosis I: management and treatment guidelines. Pediatrics. 2009;123(1):19-29. doi:10.1542/peds.2008-0416. Режим доступу: https://www.jpeds.com/article/S0022-3476(09)00675-1/fulltext
- de Ru M.H., Boelens J.J., Das A.M., Jones S.A., van der Lee J.H., Mahlaoui N. et al. (2011). Enzyme replacement therapy and/or hematopoietic stem cell transplantation at diagnosis in patients with mucopolysaccharidosis type I: results of a European consensus procedure. Orphanet journal of rare diseases, 6, 55. https://doi.org/10.1186/1750-1172-6-55. Режим доступу: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3170181/
- Ana Maria Martins, Ana Paula Dualibi, Denise Norato et al. Guidelines for the Management of Mucopolysaccharidosis Type I. SUPPLEMENT| VOLUME155, ISSUE4, SUPPLEMENT, S32-46, OCTOBER01, 2009. Режим доступу: https://www.jpeds.com/article/S0022-3476(09)00675-1/fulltext#secd52039295e1370
- Dornelles A.D., Artigalas O., da Silva A.A., Ardila D. et al. (2017). Efficacy and safety of intravenous laronidase for mucopolysaccharidosis type I: A systematic review and meta-analysis. PloS one, 12(8), e0184065. Режим доступу: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5578671/
- Jameson E., Jones S., Remmington T. Enzyme replacement therapy with laronidase (Aldurazyme®) for treating mucopolysaccharidosis type I. Cochrane Database of Systematic Reviews 2019, Issue 6. Art. No.: CD009354. DOI: 10.1002/14651858.CD009354.pub5. Режим https://www.cochranelibrary.com/cdsr/doi/10.1002/14651858.CD009354.pub5/full
Підготувала Ганна Кирпач
Тематичний номер «Діабетологія, Тиреоїдологія, Метаболічні розлади» № 2 (50), 2020 р.
СТАТТІ ЗА ТЕМОЮ Ендокринологія
Нещодавні дослідження показали, що прогноз за різних поширених захворювань, ендокринних, автоімунних розладів і навіть прогресування раку пов’язані з концентрацією вітаміну D у плазмі. Завдяки експресії гена 1α-гідроксилази (CYP27B1) клітини імунної системи (В-, Т- та антигенпрезентувальні клітини) здатні продукувати активний метаболіт кальциферол – речовину з імуномодулювальними властивостями. Рецептори до вітаміну D (vitamin D receptor, VDR) експресують на поверхні імунних клітин. Доведено зв’язок між поліморфізмом генів VDR або CYP27B1 і патогенезом автоімунних ендокринних захворювань. Метою огляду є вивчення впливу вітаміну D, наслідків його дефіциту та корисної ролі добавок із ним при деяких ендокринних розладах, які часто спостерігають у клінічній практиці. ...
Збудник COVID‑19, SARS-CoV‑2, з яким людство вперше стикнулося у 2019 р., поширився по всьому світу, заразивши мільйони людей. Сьогодні, через тягар війни та економічної нестабільності, тема COVID‑19 не сприймається так гостро, як ще кілька років тому, хоча насправді вона не втратила своєї актуальності. Саме сучасному стану проблеми COVID‑19 у світі та в Україні була присвячена доповідь директора ДУ «Інститут ендокринології та обміну речовин імені В.П. Комісаренка НАМН України», академіка Національної академії медичних наук України, члена-кореспондента НАН України, віце-президента НАМН України, президента Асоціації ендокринологів України, професора Миколи Дмитровича Тронька під час першого у 2024 р. засідання науково-освітнього проєкту «Школа ендокринолога», яке відбулося 20-24 лютого. ...
Протягом останніх 60 років метформін є найпоширенішим цукрознижувальним засобом і рекомендований як препарат першої лінії для осіб з уперше виявленим цукровим діабетом (ЦД) 2 типу. Сьогодні понад 200 млн осіб із ЦД 2 типу в усьому світі щодня застосовують метформін як монотерапію або в комбінації. Препарат усе частіше використовують для лікування гестаційного ЦД та в пацієнтів із синдромом полікістозних яєчників. ...
Двадцять восьмого лютого 2024 року виповнилося 80 років від дня народження директора ДУ «Інститут ендокринології та обміну речовин ім. В.П. Комісаренка НАМН України» (далі – Інститут), віцепрезидента НАМН України, академіка НАМН України, члена-кореспондента НАН України, заслуженого діяча науки та техніки, лауреата Державної премії України, доктора медичних наук, професора Миколи Дмитровича Тронька....