



Складна міждисциплінарна патологія: епідеміологія, діагностика і сучасні підходи до терапії хвороби Гоше в Україні
19-21 травня 2021 року в Києві відбулася науково-практична конференція «Нові можливості та невирішені питання гематології». Ця подія без перебільшення значуща як для Асоціації гематологів України, так і для всіх лікарів суміжних спеціальностей, адже багатьом із них доводиться мати справу з пацієнтами, в яких виявлено патологію крові. Під час робочої програми було заслухано доповіді на такі актуальні теми, як впровадження сучасних протоколів лікування гематологічних і онкогематологічних захворювань, реєстр і подальше амбулаторне і профілактичне лікування хворих на гемофілію; мовилося також і про практичні аспекти ведення пацієнтів з онкогематологічною патологією. Особливу увагу було приділено діагностиці й лікуванню пацієнтів із хворобою Гоше (ХГ) – вона стала темою окремого сателітного симпозіуму.
Ключові слова: глюкоцереброзидаза, хвороба Гоше, ферментний аналіз, хітотріозидаза, гепатоспленомегалія, тромбоцитопенія, кістковий криз, ферментна замісна терапія, іміглюцераза, велаглюцераза альфа, таліглюцераза альфа
Проблемним питанням діагностики ХГ присвятила свій виступ завідувач відділення гематології Київського обласного онкологічного диспансеру, голова ГО «Асоціація гематологів України», експерт робочої групи МОЗ України за напрямом «Орфанні захворювання», кандидат медичних наук Ірина Радомирівна Гартовська.
 Хвороба Гоше – найпоширеніше вроджене схильне до прогресування захворювання лізосомального накопичення, зумовлене дефектом гена, який відповідає за синтез лізосомального гідролітичного ферменту глюкоцереброзидази. У результаті дефіциту останньої в лізосомах макрофагів накопичується патологічний субстрат глюкоцереброзид. Унаслідок цього функція макрофагів порушується, і вони перетворюються на клітини Гоше. Оскільки глюкоцереброзидаза міститься в макрофагах усіх тканин, ХГ вважається поліорганною патологією, при якій у патологічний процес залучаються кістки, печінка, легені й селезінка і яка загрожує життю.
Хвороба Гоше – найпоширеніше вроджене схильне до прогресування захворювання лізосомального накопичення, зумовлене дефектом гена, який відповідає за синтез лізосомального гідролітичного ферменту глюкоцереброзидази. У результаті дефіциту останньої в лізосомах макрофагів накопичується патологічний субстрат глюкоцереброзид. Унаслідок цього функція макрофагів порушується, і вони перетворюються на клітини Гоше. Оскільки глюкоцереброзидаза міститься в макрофагах усіх тканин, ХГ вважається поліорганною патологією, при якій у патологічний процес залучаються кістки, печінка, легені й селезінка і яка загрожує життю.
Тип успадкування ХГ аутосомно-рецесивний. Захворювання з однаковою частотою спостерігається в жінок і чоловіків. На сьогодні описано понад 200 мутацій, що призводять до дефекту ферменту глюкоцереброзидази, зниження його стабільності й активності. Частота ХГ становить 1 випадок на 40-60 тис загальної популяції, а серед євреїв ашкеназі – 1 випадок на 500 осіб.
Виділяють три основних типи захворювання:
- І тип – не невропатична (вісцеральна) форма, найпоширеніша (95% випадків), має хронічний перебіг і не супроводжується неврологічними порушеннями;
- ІІ тип – гостра невропатична форма, яку діагностують у дітей віком 1-2 роки та яка характеризується злоякісним перебігом і високим рівнем смертності;
- ІІІ тип – підгостра невропатична форма, яку діагностують у підлітків.
Станом на 2020 рік в Україні перебуває на обліку 58 пацієнтів із ХГ, із них – 4 пацієнти з ІІІ типом захворювання.
Таким чином, частота ХГ І типу в Україні в 14 разів, а ІІІ типу – в 100 разів нижча, ніж загалом у світі. Подібна картина, імовірно, зумовлена гіподіагностикою цього стану, що пов’язано з недостатньою обізнаністю медичної спільноти щодо цього орфанного захворювання. Це стосується переважно педіатрів, сімейних лікарів, гастроентерологів і дитячих і дорослих гематологів, до яких зазвичай звертаються пацієнти.
Крім складнощів у діагностиці проблемними залишаються питання забезпечення препаратами ферментної замісної терапії (ФЗТ) і переведення з одного препарату на інший. Забезпеченість пацієнтів препаратами замісної терапії складає приблизно 70%. Сьогодні на фармацевтичному ринку України присутні три препарати замісної терапії ХГ: іміглюцераза, велаглюцераза і таліглюцераза. Зауважимо, що всі три препарати є оригінальними і не поступаються один одному в ефективності і безпеці. Переведення пацієнтів на економічно вигідніший із засобів дасть можливість забезпечити більшу частку пацієнтів, що отримують ФЗТ, а також вивільнить кошти для лікування інших орфанних захворювань. На жаль, часто лікарі бояться переводити пацієнтів з іміглюцерази на велаглюцеразу альфа чи таліглюцеразу альфа, незважаючи на позитивний досвід таких країн, як Австралія, Сербія, Албанія і Канада.
Із власним досвідом ведення пацієнтів із ХГ від діагностики до лікування на прикладі двох клінічних випадків поділилася з учасниками конференції завідувач відділення інтенсивної хіміотерапії ДУ «Інститут гематології та трансфузіології АМН України» (м. Київ), доктор медичних наук, професор Світлана Олексіївна Сівкович.
 Складнощі діагностики ХГ, крім невисокої частоти патології в популяції, зумовлені її мультисимптомністю і безліччю захворювань з аналогічною симптоматикою, зокрема таких як злоякісні новоутворення, онкологічні захворювання крові (лейкемія, лімфома, множинна мієлома, хронічна гранулоцитарна лейкемія), аутоімунні захворювання, цироз печінки та інші.
Складнощі діагностики ХГ, крім невисокої частоти патології в популяції, зумовлені її мультисимптомністю і безліччю захворювань з аналогічною симптоматикою, зокрема таких як злоякісні новоутворення, онкологічні захворювання крові (лейкемія, лімфома, множинна мієлома, хронічна гранулоцитарна лейкемія), аутоімунні захворювання, цироз печінки та інші.
В основі моделі ведення пацієнтів із ХГ лежить своєчасна діагностика патології до розвитку незворотних ускладнень. Початкові прояви ХГ І типу характеризуються такими гематологічними ознаками: спленомегалія, анемія, тромбоцитопенія і схильність до кровотеч. Часто відповідні скарги і зміни в клінічному аналізі крові стають приводом для направлення пацієнта до гематолога.
Основні клінічні симптоми ХГ І типу зазвичай представлені затримкою фізичного і статевого розвитку в дітей, гепатоспленомегалією, астенічним і геморагічним синдромами, болем у кістках, у тому числі кістковими кризами. Розвиток ускладнень призводить до порушення рухливості в суглобах (унаслідок розвитку асептичного некрозу) і патологічних переломів.
Золотим стандартом лабораторної діагностики ХГ є ферментний аналіз, за допомогою якого визначають активність глюкоцереброзидази в лейкоцитах крові або фібробластах шкіри. У разі ХГ результат дослідження становить менше 30% від нормальної активності ферменту. Ще одним ферментом, визначення активності якого є важливим для встановлення ступеня тяжкості хвороби і результативності лікування, є хітотріозидаза. Пацієнтам із підозрою на ХГ також потрібні молекулярний аналіз і консультація генетика.
Зміни в загальному аналізі крові (ЗАК), які можуть вказувати на наявність ХГ: анемія; лейкопенія і тромбоцитопенія різного ступеня тяжкості. Біохімічний аналіз крові (БАК) дає уявлення про рівні трансферину і феритину сироватки крові.
Діагностично важливою ознакою при ХГ є гіперферитинемія, яку спостерігають у 87% пацієнтів із І типом захворювання.
Випадковою знахідкою при біопсії кісткового мозку можуть стати клітини Гоше. Але варто пам’ятати, що при різних патологічних станах можуть бути виявлені псевдоклітини Гоше. Такими станами є множинна мієлома, мієлодисплазія і мієлодиспластичні синдроми, хронічна мієлоїдна лейкемія, туберкульоз легень, мікобактеріоз і серпоподібноклітинна анемія. Це дає підстави вважати зазначений метод діагностики недостатньо специфічним і чутливим. Утім біопсія кісткового мозку може бути показана пацієнтам із ХГ і підозрою на наявність супутньої гематологічної патології.
Після підтвердження діагнозу варто зосередитися на комплексній оцінці всіх аспектів захворювання для встановлення вихідних характеристик, визначення типу ФЗТ, розробки індивідуальних терапевтичних цілей і стратегії моніторингу стану здоров’я хворого. Під час прийняття рішення стосовно лікування треба оцінити тяжкість захворювання і показники якості життя.
Особливо важливою є ретельна оцінка кісток скелету, оскільки можуть несподівано виникати незворотні ускладнення, що призводять до інвалідизації.
Наступним кроком після оцінки тяжкості захворювання є підбір відповідної ФЗТ. Результати Кокранівського огляду показали, що впродовж першого року ФЗТ різні рекомбінантні препарати є біоподібними і зіставними щодо безпеки й ефективності (табл. 1).
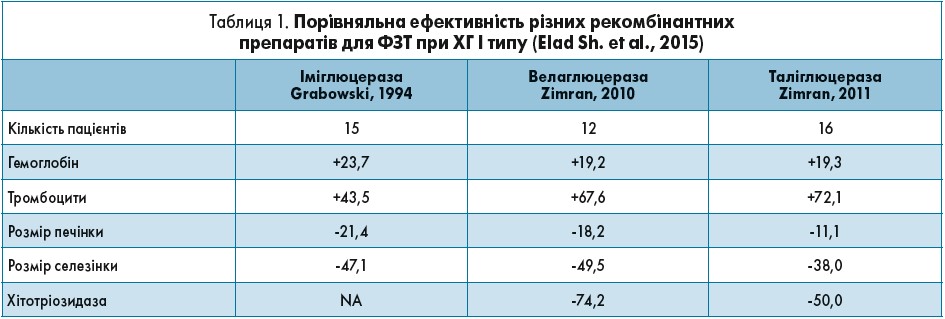
Таким чином, лікування пацієнтів із ХГ доцільно проводити будь-яким доступним препаратом для ФЗТ. Лікар має забезпечити пацієнтові:
- доступ до ФЗТ і безперервного лікування;
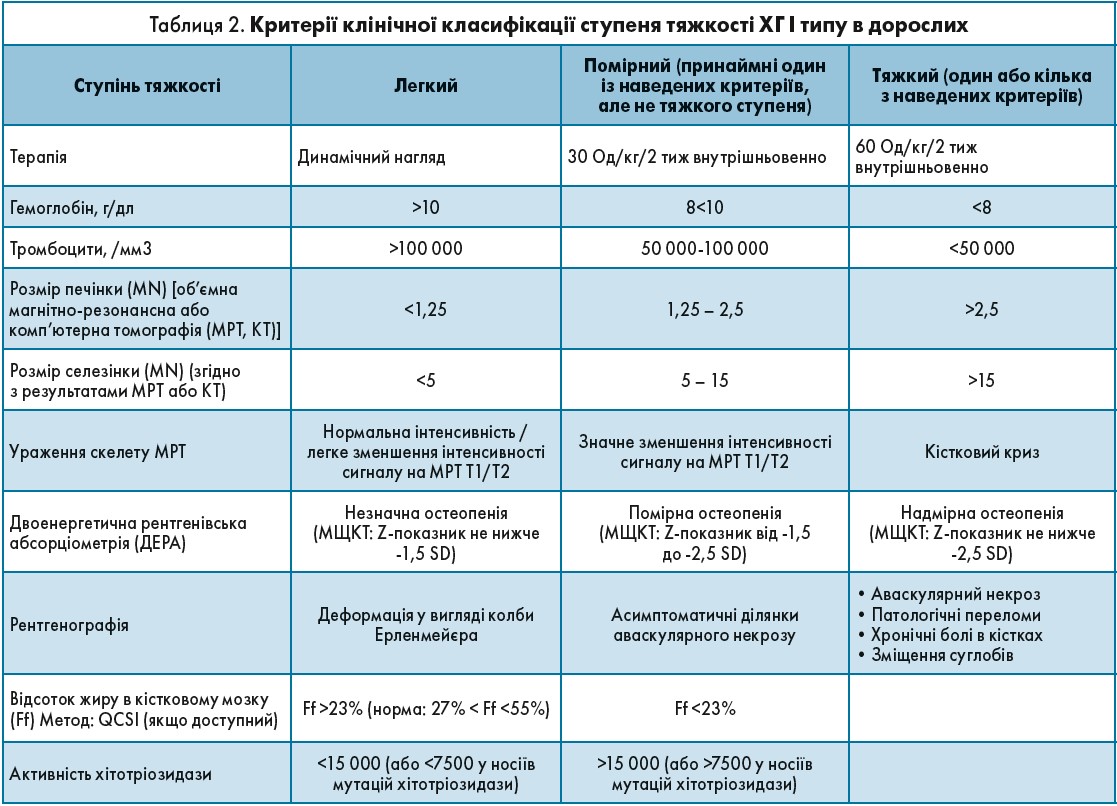
- необхідну дозу засобу ФЗТ відповідно до тяжкості перебігу захворювання згідно з критеріями клінічної класифікації (табл. 2);
- лікування супутніх захворювань для уникнення подальших ускладнень ХГ;
- покращення якості життя.
Для кращого розуміння практичних аспектів ведення пацієнтів із підозрою на ХГ розглянемо два клінічні випадки.
Клінічний випадок 1
Пацієнт, 75 років. Уперше діагноз ХГ встановлено 1998 року, підтверджено в Національній дитячій спеціалізованій лікарні (НДСЛ) «ОХМАТДИТ» 2011 і 2018 року:
- ферментний аналіз – зниження активності бета-глюкозидази до 2,7 ммоль/год/мг (норма ≥4,1 ммоль/год/мг);
- молекулярна діагностика ХГ – виявлено два гетерозиготних патогенних варіанти гена GBA, аутосомно-рецесивний тип успадкування;
- концентрація глюкозилцераміду (lyso-GB1) – 762,0 нг/мл (норма ≤10,0 нг/мл).
В анамнезі: резекція селезінки в 1998 році; гепатомегалія, деформівний артроз правого кульшового суглоба (2006 рік), ІІІ група інвалідності. Під час огляду скаржився на слабкість, втому, біль у кістках, кульшових суглобах, лівому коліні.
МРТ кульшових суглобів виявила ознаки правобічного коксартрозу ІІІ ступеня з дифузними патологічними змінами структури кісткового мозку стегнової кістки та ацетабулярної западини праворуч, які, ураховуючи анамнестичні дані, можуть відповідати остеонекрозу на тлі ХГ. МР-ознаки помірного ексудативно-проліферативного синовіїту правого кульшового суглоба з вільними хондромними тілами; лівобічного коксартрозу ІІ ступеня на тлі феморо-ацетабулярного імпінджменту за змішаним кулачково-пінцетним типом, лівобічного зовнішнього синдрому клацання стегна. МР-ознаки вогнищ остеонекрозу в кістках таза зазначеної локалізації, стрес-реактивних змін крижової кістки. Системний остеопороз.
З 2019 року почав ФЗТ таліглюцеразою альфа в дозі 60 Од/кг 1 раз на 2 тижні. Періодично відбувалися перерви в лікуванні, які супроводжувалися погіршенням стану хворого. З грудня 2020 року отримує ФЗТ на постійній основі. На жаль, ФЗТ розпочато після розвитку незворотних кісткових змін, тому повний регрес симптомів неможливий. Утім застосування ФЗТ навіть на цьому етапі захворювання запобігає подальшому прогресуванню ускладнень і підтримує якість життя хворого.
Клінічний випадок 2
Пацієнт, 72 роки. Уперше діагноз ХГ встановлений 2011 року у зв’язку зі спленомегалією, яка супроводжувалася синдромом гіперспленізму, тромбоцитопенією (мінімальний рівень – 30 тис). Діагноз підтверджений у НДСЛ «ОХМАТДИТ» 2019 року:
- проведена стернальна пункція – виявлені клітини Гоше;
- ферментний аналіз – зниження активності бета-глюкозидази до 2,2 нмоль/год/мг;
- молекулярна діагностика ХГ – виявлена мутація Asn409Ser у гені GBA в гомозиготному стані.
Основні скарги: біль в епігастрії, збільшення живота, носові кровотечі, підвищена стомлюваність.
ЗАК на момент призначення ФЗТ: тромбоцити – 39 тис/мм3, швидкість осідання еритроцитів (ШОЕ) 56 мм/год.
УЗД органів черевної порожнини (ОЧП): Печінка і селезінка рівномірно збільшені. Структура селезінки дифузно неоднорідна, виявляються гіперехогенні вогнища діаметром 28-48 мм.
Денситометрія – МЩКТ відповідає помірній остеопенії.
ФЗТ розпочато наприкінці 2020 року. Через 4 міс терапії стан пацієнта значуще покращав. А саме: усі скарги зникли; зменшилися розміри печінки і селезінки; нормалізувалися показники крові; ознак ураження кістково-суглобової системи не виявлено.
Таліглюцераза альфа, яку використовували для лікування в обох клінічних випадках, – це перший схвалений FDA (Управління із санітарного нагляду за якістю харчових продуктів і медикаментів США) рекомбінантний білок, експресований у рослинних клітинах, рекомендований для лікування пацієнтів із підтвердженим діагнозом ХГ І типу. Таліглюцераза альфа схвалена у більш як 10 країнах світу і дозволена для використання в дітей віком від 4 років. Препарат виробляється в одноразових біореакторах, він захищений природним бар’єром від вірусного забруднення і не містить компонентів ссавців (Fоx J.L. et al., 2012; Shaaltiel Y. et al., 2007).
Безпека й ефективність таліглюцерази альфа в дозах 30 Од/кг і 60 Од/кг були продемонстровані в базовому 9-місячному подвійному сліпому рандомізованому дослідженні в паралельних групах ІІІ фази (РВ‑06-001) у дорослих раніше нелікованих пацієнтів із ХГ (Zimran A. et al., 2012). Пацієнтам, які завершили дослідження РВ‑06-001, було запропоновано взяти участь у багатоцентровому подвійному сліпому рандомізованому дослідженні ІІІ фази з 2 рівнями дозування: 30 або 60 Од/кг внутрішньовенно через тиждень РВ‑06-003. Оцінку ефективності проводили за розмірами селезінки й печінки, кількістю тромбоцитів, концентрацією гемоглобіну та активністю хітотріозидази. Безпеку оцінювали за частотою небажаних явищ (НЯ) і титром антитіл до таліглюцерази альфа.
Результати, отримані через 36 міс терапії таліглюцеразою альфа, відображено на рисунках 1-3.
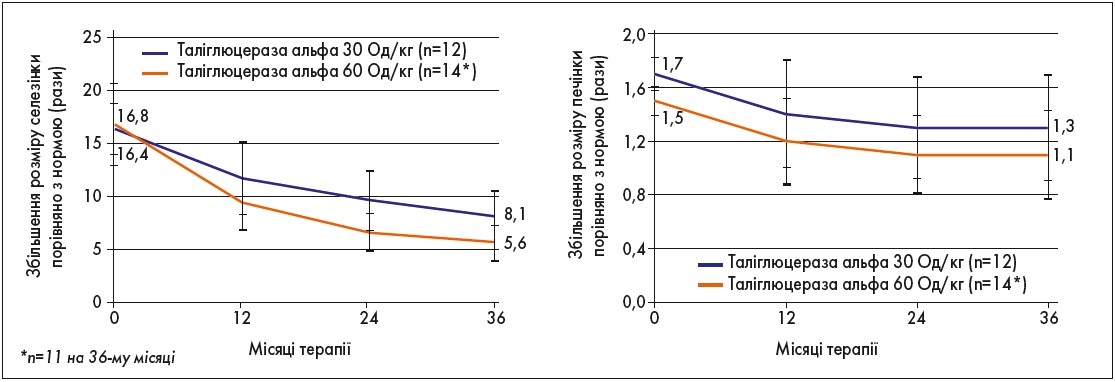
Рис. 1. Зменшення розмірів селезінки й печінки на тлі 36-місячної ФЗТ таліглюцеразою альфа (РВ‑06-001; РВ‑06-003)
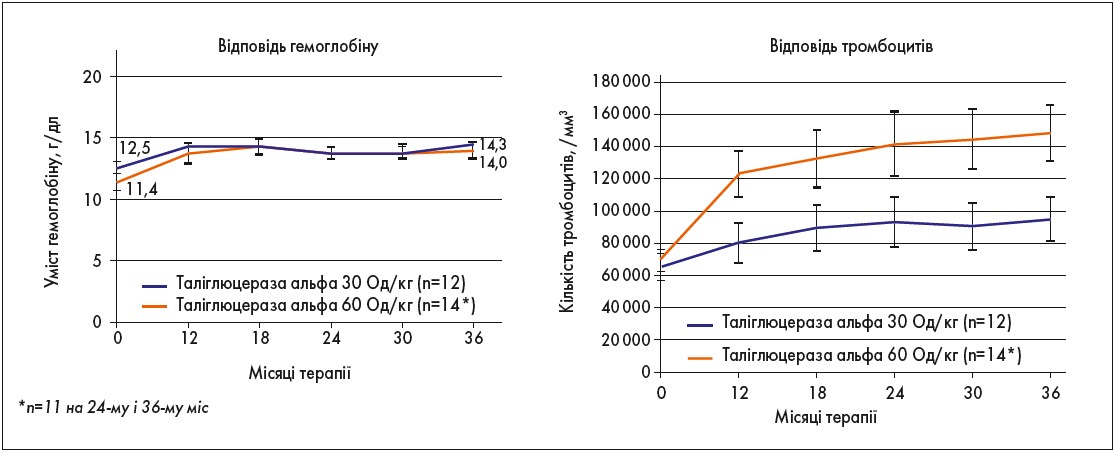
Рис. 2. Нормалізація гематологічних показників на тлі 36-місячної ФЗТ таліглюцеразою альфа (РВ‑06-001; РВ‑06-003)
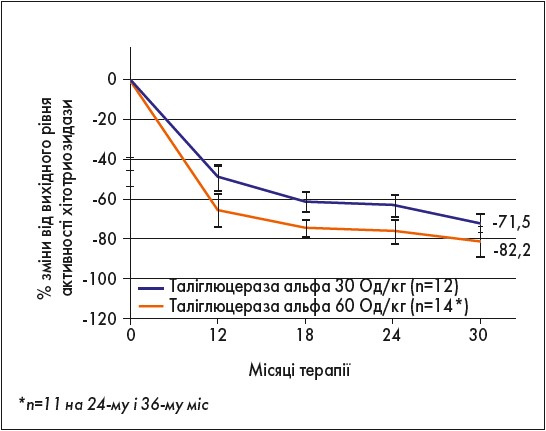
Рис. 3. Активність хітотріозидази на тлі 36-місячної ФЗТ таліглюцеразою альфа (РВ‑06-001; РВ‑06-003)
Отже, дослідження РВ‑06-001 і РВ‑06-003 показали, що лікування таліглюцеразою альфа впродовж 36 міс у дорослих пацієнтів із ХГ, які до цього не отримували ФЗТ, забезпечило тривале поліпшення всіх фізикальних і лабораторно-інструментальних параметрів тяжкості захворюванна, зокрема:
- нормалізацію кількості тромбоцитів і гемоглобіну;
- зменшення об’ємів селезінки і печінки;
- зниження активності хітотріозидази;
- збільшення жовтого кісткового мозку, що відображає кліренс клітин Гоше.
Усі НЯ, пов’язані з лікуванням, були слабкими або помірними і швидко зникали.
Із доповіддю «Від симптому до діагнозу – діагностичний алгоритм хвороби Гоше» виступила лікар-педіатр центру орфанних захворювань НДСЛ «ОХМАТДИТ» МОЗ України Яна Ігорівна Дороніна.
 Хвороба Гоше – це патологія, яка характеризується вираженою гетерогенністю клінічної картини і прогресуючим перебігом (Mistry P.K. et al., 2012; Brady R.O. et al., 1997). Поряд із гепато- і спленомегалією і гематологічними змінами у вигляді цитопенії різного ступеня вираженості ХГ в дітей часто маніфестує кістковими проявами. Саме на цьому аспекті зупинимося детальніше.
Хвороба Гоше – це патологія, яка характеризується вираженою гетерогенністю клінічної картини і прогресуючим перебігом (Mistry P.K. et al., 2012; Brady R.O. et al., 1997). Поряд із гепато- і спленомегалією і гематологічними змінами у вигляді цитопенії різного ступеня вираженості ХГ в дітей часто маніфестує кістковими проявами. Саме на цьому аспекті зупинимося детальніше.
Ураження кістково-суглобової системи, які є основним фактором інвалідизації хворих, спостерігають у більш як 90% пацієнтів із ХГ (Kaplan P. et al., 2013). Найчастіше ураження зазнають трубчасті кістки. В основі патогенезу лежить інфільтрація кісткового мозку клітинами Гоше, яка спричиняє деформацію кісток у вигляді колб Ерленмеєра через порушення процесів ремоделювання кістки.
Скелетні ураження при ХГ найчастіше проявляються больовим синдромом. Біль неспецифічний, ниючий, може бути гострим або хронічним, епізодичним чи постійним. Патогномонічними при ХГ є кісткові кризи, які можуть імітувати гострий остеомієліт і супроводжуються сильним болем, який часто потребує прийому наркотичних аналгетиків; лихоманкою, лейкоцитозом.
При природному перебігу ХГ без застосування ФЗТ згодом розвиваються скелетні ускладнення, які прогресують і значно знижують якість життя пацієнтів (Marcucci G. et al., 2014):
- хронічна ішемія кістки з кістковими інфарктами;
- остеонекроз (локалізована загибель кістки), який найчастіше вражає головку і шию стегна, проксимальні відділи плечової і великогомілкової кістки і хребці, що супроводжується сильним болем і призводить до інвалідизації;
- літичні ураження – невеликі ерозії внаслідок локалізованого руйнування кістки;
- остеосклероз – аномальне затвердіння або збільшення щільності кістки через гіперактивне запалення;
- колапс тіла хребця з відростанням периферичних клітин, «провисанням», формуванням Н-подібної форми. Призводить до візуального дефекту і/або перелому хребця;
- руйнування кісток або суглобів, які можуть проявлятися у вигляді інтенсивного, часто хронічного, болю, інвалідизації і/або деформації кісток; може охоплювати патологічні переломи, колапс.
Єдиним способом уникнення всіх названих ускладнень і ранньої інвалідизації є вчасне встановлення діагнозу і призначення ФЗТ.
Методи клінічного і лабораторного обстеження за підозри/наявності ХГ:
1. ЗАК для виявлення цитопенії.
2. БАК, де найчастіші зміни представлені підвищенням рівня феритину, рідко АЛТ, АСТ і білірубіну.
3. Коагулограма, в якій може спостерігатися збільшення протромбінового часу.
4. УЗД і МРТ печінки і селезінки, які дають можливість виявити вогнищеві ураження і визначити початковий об’єм органів, що необхідно для подальшого контролю ефективності ФЗТ.
5. Денситометрія і МРТ – найбільш чутливі методи, на відміну від рентгенографії допомагають виявити ураження кісток (остеопенію, інфільтрацію кісткового мозку) на ранніх стадіях.
6. Дослідження функції нервової системи, зокрема дослідження руху очей, додаткові нейроофтальмологічні тести, аналіз периферійного слуху, томографія мозку, електроенцефалографія і нейропсихометрія.
7. Допплер-ехокардіографія в пацієнтів після спленектомії.
8. Морфологічне дослідження кісткового мозку на сьогодні не є обов’язковим для діагностики ХГ, ураховуючи інвазивність методу і високу частоту хибно-негативних результатів, однак він допомагає віддиференціювати ХГ від інших лімфопроліферативних захворювань.
9. Специфічна діагностика ХГ, яка полягає у визначенні активності бета-глюкозидази (зниження до 5% вказує на ХГ) і мутації в гені GBA.
Для скринінгу ХГ може використовуватися метод «сухої плями крові», яку оцінюють у лабораторії ARCHIMED (Австрія) або CENTOGENE (Німеччина). Водночас варто зауважити, що результат зазначеного аналізу не може бути підставою для встановлення діагнозу і має бути підтверджений у лабораторії центру орфанних захворювань НДСЛ «ОХМАТДИТ» вимірюванням активності бета-глюкозидази в лейкоцитах цільної крові.
На жаль, рідкісність патології, пізня маніфестація, різнорідність і неспецифічність проявів, а також недостатня обізнаність щодо цієї хвороби серед лікарів різного профілю часто призводять до пізньої діагностики ХГ (Mehta A. et al., 2015). Згідно з власними спостереженнями центру орфанних захворювань НДСЛ «ОХМАТДИТ», діапазон появи початкових проявів захворювання коливається від 1 до 45 років, із середнім віком маніфестації 8 років. Затримка встановлення діагнозу становить 3 роки в дітей і до 8 років у дорослих. Більш як у 35% хворих діагноз встановлюють невірно.
Наслідки невчасної діагностики ХГ і призначення ФЗТ:
- неухильне прогресування захворювання;
- підвищений ризик виникнення аваскулярного некрозу;
- неконтрольована кровотеча, особливо після хірургічних втручань;
- легенева гіпертензія;
- затримка росту;
- фатальна септицемія, передусім у пацієнтів після спленектомії;
- патологічні переломи / кісткові інфаркти;
- непотрібні/невідповідні обстеження;
- неадекватна, а іноді й небезпечна терапія.
З іншого боку, вчасно розпочата ФЗТ дає можливість мінімізувати прояви захворювання і забезпечити високу якість життя пацієнтам із ХГ. В Україні призначення ФЗТ здійснюють комісійно, відповідно до наказу МОЗ України від 12.05.2015 № 50 «Про затвердження Положення про комісію для визначення необхідності в призначенні, відміні, перерозподілі лікарських засобів та відповідних харчових продуктів для спеціального дієтичного споживання, що закуповуються за рахунок коштів державного бюджету, а також інших джерел, не заборонених законодавством, у тому числі гуманітарної допомоги, громадянам, які страждають на рідкісні (орфанні) захворювання».
Важливо пам’ятати, що в разі переривання лікування або неоптимального режиму терапії можливе прогресування хвороби кісток або їх незворотне пошкодження. Саме тому внутрішньовенні інфузії засобів ФЗТ виконують регулярно, а дозу підбирають індивідуально, відповідно до клінічного стану пацієнта. Для контролю адекватності терапії на постійній основі (кожні 6-12 міс) фахівці різних спеціальностей проводять комплексний моніторинг усіх параметрів захворювання.
У педіатрів і дитячих гематологів має бути особлива насторога щодо ХГ, адже часто захворювання маніфестує саме в дитячому віці. Необхідно розуміти ускладнення, з якими пов’язане відстрочення ФЗТ, і вчасно направляти пацієнтів на специфічну діагностику і подальше лікування.
Детальніше про особливості лабораторної діагностики ХГ в Україні розповіла біолог вищої категорії лабораторії медичної генетики центру орфанних захворювань НДСЛ «ОХМАТДИТ» Світлана Володимирівна Кормоз. 
Алгоритм діагностики ХГ складається з трьох послідовних кроків.
1. Визначення активності глюкоцереброзидази.
- Для цього використовують два основні біохімічні методи:
- флюорометричне визначення активності бета-глюкоцереброзидази в сухій плямі крові (DBS) і цільній крові з ЕДТА (лейкоцити);
- тандемна мас-спектрометрія LC-MS/MS, матеріалом для якої є суха пляма крові.
2. Виявлення вторинних біохімічних маркерів, які свідчать про накопичення субстрату в клітинах.
Для цього використовують:
- флюорометричне визначення активності хітотріозидази в плазмі крові (Wallac 1402 Victor Multi Label Counter);
- тандемну мас-спектрометрію LC-MS/MS для визначення рівня глюкозилцераміду (Lyso-Gb1) в сухій плямі крові.
3. Виявлення мутацій у гені GBA.
За даними HGMD® (База даних мутацій у генах людини – Human Gene Mutation Database) 2020, описано понад 540 мутацій у гені GBA, які пов’язують із ХГ (Hruska K.S. et al., 2008). З 2017 року в «ОХМАТДИТ» стартував проєкт із визначення генетичної природи ХГ в пацієнтів, діагноз яким було встановлено з 2001 року за результатами комплексу клінічних, морфологічних і біохімічних даних.
Молекулярно-генетичні методи діагностики охоплюють:
- метод «гніздової» полімеразно-ланцюгової реакції (ПЛР) для отримання фрагментів послідовності функціонального гена GBA (AB SimpliAmpTM Thermal Cycler);
- поліморфізм довжини рестрикційних фрагментів (ПДРФ) – аналіз для визначення мажорних міссенс-замін у гені GBA (Xhol і Mspl відповідно) (Shimadzu MultiNA microchip electrophoresis system);
- пряме поекзонне сиквенування за Сенгером для ідентифікації рідкісних перебудов (ABI Prism 3130).
Потрібно акцентувати увагу на вимогах щодо забору, зберігання і транспортування біологічного матеріалу, який використовують для діагностики ХГ (табл. 3).

Під час забору сухих плям крові необхідно дотримуватися певних правил:
- У немовлят до 12 міс кров беруть із п’ятки, після 12 міс – із пальця.
- Місце майбутнього проколу треба зігріти, промасажувати, протерти антисептиком, після чого висушити сухою стерильною серветкою.
- Після проколу не треба видаляти першу краплю.
- Заборонено здавлювати п’ятку/палець для пришвидшення току крові.
- Кожне коло бланку треба просочувати одномоментно, однією краплею крові, наскрізь.
- Не можна торкатися бланком шкіри.
- Категорично заборонено торкатися або здавлювати пляму крові.
Найпоширенішими помилками під час забору сухих плям є недостатня кількість зразка для дослідження, здавлювання зразка, погане висушування, розведення або забруднення зразка і повторне нашарування крові. Зразки, перенасичені кров’ю, і забір не капілярної, а венозної крові (наявність сивороткових кілець) також не можна використовувати для дослідження.
Варто зауважити, що на активність ферментів у сухих плямах крові значуще впливають фактори довкілля, зокрема термін і температура зберігання, умови доставки (температура і вологість), спосіб і умови висушування і кількість лейкоцитів і гематокрит.
Таким чином, визначення активності ферментів у сухій плазмі крові – це простий малоінвазивний спосіб біохімічної діагностики ХГ, який потребує малого об’єму крові і характеризується мінімальним ризиком псування внаслідок гемолізу або бактеріальної контамінації, а також великим терміном придатності. Недоліками методу є його залежність від правильного забору, умов доставки і зберігання. Крім того, метод характеризується порівняно високою частотою хибнопозитивних результатів при ферментодіагностиці лізосомальних хвороб накопичення.
Власні дані лабораторії центру орфанних захворювань НДСЛ «ОХМАТДИТ» виявили хибнопозитивні результати DBS-скринінгу в 9,3% (15/160) пацієнтів при діагностиці ХГ, 1,6% (8/502) – при хворобі Помпе, 1% – при хворобі Фабрі (2/200) і 1,4% (2/146) – при мукополісахаридозі І типу.
Отже, ферментативний аналіз на DBS – це скринінгове дослідження, яке потребує обов’язкового підтвердження на цільній крові в лабораторії центру орфанних захворювань НДСЛ «ОХМАТДИТ».
Наприкінці сателітного симпозіуму лікар-гематолог вищої категорії, завідувач консультативної поліклініки, ДУ «Інститут патології крові та трансфузійної медицини НАМН України» Ірина Михайлівна Юрчишак на прикладі клінічного випадку продемонструвала, яким непростим може бути діагностичний шлях пацієнтів із ХГ.
Клінічний випадок 3
Пацієнтка, 29 років. 05.07.2017 року вперше звернулася на консультацію до лікаря-гематолога консультативної поліклініки з приводу спленомегалії (+8 см) і змін у ЗАК (анемія: гемоглобін 75 г/л, тромбоцитопенія 80 тис/мм3). 29.11.2017 року хворій встановили діагноз – ідіопатичний мієлофіброз (ІМФ). Жінку було взято на диспансерний облік у консультативну поліклініку ДУ «Інститут патології крові та трансфузійної медицини НАМН України».
Із 28.12.17 по 05.01.18 рік хвора вперше лікувалася стаціонарно в гематологічному відділенні 5-ї МКЛ з діагнозом: ІМФ, розгорнута стадія. Спленомегалія.
При поступленні в стаціонар пацієнтка скаржилася на загальну слабкість, запаморочення, утомлюваність, важкість у лівому підребер’ї. Погіршення самопочуття зазначає з 2017 року. На основі трепан-біопсії кісткового мозку діагностований мієлофіброз. При огляді на момент звернення – селезінка +8 см, чутлива при пальпації. ЗАК при госпіталізації: гемоглобін – 75 г/л, лейкоцити – 2,1×109/л, тромбоцити – 80 тис/мм3. Після лікування (гідроксисечовина, алопуринол, гемотрансфузії, глюкокортикостероїди (ГКС), гепатопротектори) стан хворої покращився. ЗАК при виписці: гемоглобін – 137 г/л, еритроцити – 4,9×1012/л, лейкоцити – 5,7×109/л, тромбоцити – 317 тис/мм3. Рекомендований повторний огляд гематолога через 2 тижні.
Наступна зустріч із пацієнткою відбулася майже через рік. Через погіршення стану вона знову була госпіталізована і отримувала аналогічне лікування із 07.12.18 по 12.12.18 рік. Виписана з покращенням, після чого на контроль до гематолога не з’являлася.
У лютому 2020 року стан хворої знову погіршився, однак, у зв’язку з епідемією COVID‑19, жінка змогла приїхати на прийом лише 31.07.2020 року. Скарги на момент звернення: швидка втомлюваність, постійна загальна слабкість, дискомфорт і відчуття тиску в лівій половині живота, періодична поява синців на тулубі, худорлявість, болі в кістках після бігу (до 5 км).
УЗД ОЧП за 11.06.2020 рік: гепатолієнальний синдром (розміри селезінки 233×89 мм), сонографічні ознаки портальної гіпертензії.
Ураховуючи бажання материнства, вік пацієнтки, результати ЗАК, БАК і гігантську селезінку, було запропоновано провести подальше дообстеження, а саме:
- трепан-біопсію клубової кістки (для встановлення ступеня фіброзу);
- визначення рівнів еритропоетину, феритину, сироваткового заліза;
- ПЛР до вірусів гепатиту В і С (кров, якісно).
Під час детального збору анамнезу виявилось, що в дитинстві в пацієнтки були часті скарги на втомлюваність, болі в кістках, часті носові кровотечі, синячковість, які батьки пояснювали швидким ростом і надмірною активністю дитини.
Трепанобіоптат. Трепанобіоптат клітинний. Гранулоцитарний паросток представлений усіма перехідними формами, омолоджений до мієлоцитів. Нормобластичний тип еритропоезу, представлений нормоцитами. Мегакаріоцити поодинокі, діяльні.
Рівень заліза сироватки в нормі, тоді як показники феритину та еритропоетину – виражено підвищені (217 нг/мл і 85,5 мОд/мл відповідно). Вірусні гепатити не виявлено.
Таким чином, даних, які б свідчили про наявність мієлофіброзу, виявлено не було. Було висловлено підозру на ХГ і зроблено повторний перегляд трепанобіоптату за 2017 рік, з акцентом на пошук специфічних клітин Гоше. Паралельно була забрана кров методом DBS і направлена на безкоштовну діагностику в Німеччину.
Детальний перегляд трепанобіоптату виявив таке: у досліджуваному матеріалі, серед збереженого кісткового мозку, в якому визначаються клітини еритроїдного, гранулоцитарного і мегакаріоцитарного паростків кровотворення, візуалізуються масивні ділянки проліфератів патологічних гістіоцитів зі світлою еозинофільною плазмою з вираженою шаруватістю (феномен «м’ятого папірусного паперу») і невеликими округлими базофільними ядрами. Морфологія процесу найбільше відповідає ХГ (глюкозилцерамідному ліпідозу).
Після цього була проведена специфічна діагностика ХГ, яка виявила:
- активність бета-глюкозидази 0,7 нмоль/год/мл (норма 5,1-9,5 нмоль/год/мл);
- активність хітотріазидази – 9017 нмоль/год/мл (норма 0-159 нмоль/год/мл);
- молекулярно-генетичне дослідження гена GBA виявило мутацію p.(Asn409Ser) в гомозиготному стані.
Відповідно до отриманих результатів обстежень у листопаді 2020 року діагноз «ідіопатичний мієлофіброз» було змінено на «хворобу Гоше». Після цього пацієнтку було включено на заявку МОЗ для забезпечення ФЗТ. До поставки препарату за кошти держбюджету хвора отримувала терапію велаглюцеразою альфа, яка була надана гуманітарною програмою.
Крім того, пацієнтці рекомендований контроль аналізів:
- рівня хітотріазидази – 1 р./рік;
- ЗАК, БАК, електролітів крові – 1 р./6 міс;
- УЗД ОЧП 1 р./6-12 міс, у разі зміни структури –
- КТ/МРТ;
- МРТ кісток тулуба і кінцівок;
- денситометрія – 1 р./рік.
Прогноз хвороби для пацієнтки сприятливий. Можна очікувати нормалізації показників периферичної крові та до 1-2 років – розмірів селезінки. Крім того, ХГ не є протипоказанням для настання вагітності. Але планувати її доцільно після досягнення цілей лікування ХГ. Питання про продовження ФЗТ під час вагітності і годування груддю вирішується в індивідуальному порядку з урахуванням стану пацієнтки і її прихильності до лікування. Спосіб родорозрішення визначають за акушерськими показаннями з урахуванням наявності цитопенії і стану системи гемостазу (Paterman A.H. et al., 2007).
Активний обмін думками в рамках сателітного симпозіуму продемонстрував високий освітній і професійний рівень представлених доповідей. Спікери доступно і достатньо ґрунтовно окреслили головні практичні аспекти ведення хворих із підозрою на ХГ і зробили це орфанне захворювання зрозумілішим як для гематологів, так і для педіатрів і лікарів загальної практики – сімейної медицини, які найчастіше першими стикаються з пацієнтами, в яких наявні певні симптоми цієї рідкісної патології.
Підготувала Ганна Кирпач
Тематичний номер «Діабетологія, Тиреоїдологія, Метаболічні розлади» № 2 (54) 2021 р.
СТАТТІ ЗА ТЕМОЮ Ендокринологія
21 березня в рамках II Міжнародної школи «Сучасний лікар: від теорії до практики» професор кафедри ендокринології Львівського національного медичного університету ім. Данила Галицького, лікар-ендокринолог вищої категорії, доктор медичних наук Вікторія Олександрівна Сергієнко представила доповідь, присвячену хронічним ускладненням цукрового діабету (ЦД). Зокрема, було акцентовано увагу на причинах розвитку діабетичної полінейропатії (ДП), розглянуто клінічні варіанти цього ускладнення, діагностичні підходи та основні принципи лікування. Пропонуємо огляд цієї доповіді у форматі «запитання – відповідь»....
Останніми десятиліттями в усьому світі спостерігалося значне зростання поширеності цукрового діабету (ЦД), що зумовило серйозні наслідки стосовно якості життя населення, а також спричинило певний тягар для системи охорони здоров’я та економічні витрати [1]. За даними Діабетичного атласу Міжнародної діабетичної федерації (International Diabetes Federation Diabetes Atlas), у 2021 р. ≈537 млн людей мали ЦД і, за прогнозами, до 2045 р. цей показник досягне 783 млн [2]. Значна захворюваність і підвищена смертність асоційовані з пов’язаними із ЦД макросудинними (інфаркт міокарда, інсульт) і мікросудинними (сліпота, ниркова недостатність, ампутації) ускладненнями [3]....
За визначенням Всесвітньої організації охорони здоров’я, цукровий діабет (ЦД) – це група метаболічних розладів, що характеризуються гіперглікемією, яка є наслідком дефектів секреції інсуліну, дії інсуліну або обох цих чинників. За останні 15 років поширеність діабету зросла в усьому світі (Guariguata et al., 2014). Згідно з даними Diabetes Atlas (IDF), глобальна поширеність діабету серед осіб віком 20-79 років становила 10,5% (536,6 млн у 2021 році; очікується, що вона зросте до 12,2% (783,2 млн у 2045 році (Sun et al., 2022). Наразі триває Програма профілактики діабету (ППД), метою якої є визначити, які підходи до зниження інсулінорезистентності (ІР) можуть допомогти в створенні профілактичних заходів ЦД 2 типу (The Diabetes Prevention Program (DPP), 2002). У цьому світлі визначення впливу вітаміну D на розвиток ЦД є актуальним питанням....
Внутрішній біологічний годинник людини тісно та двоспрямовано пов’язаний зі стресовою системою. Критична втрата гармонійного часового порядку на різних рівнях організації може вплинути на фундаментальні властивості нейроендокринної, імунної та вегетативної систем, що спричиняє порушення біоповедінкових адаптаційних механізмів із підвищеною чутливістю до стресу й уразливості. Поєднання декількох хвороб зумовлює двоспрямованість патофізіологічних змін....